Handling a dual-occupancy ligand with autoBUSTER
If you are only interested in occupancy refinement, and not in fitting things in coot from scratch, jump straight to Automatic occupancy refinement.
This example is for a ligand which fits in two similar conformations, distinguished
by a rotation of a benzene ring with two substituents. It is structure 1pmq from
the PDB, done by Giovanna Scapin of Merck; the protein is the JNK3 kinase.
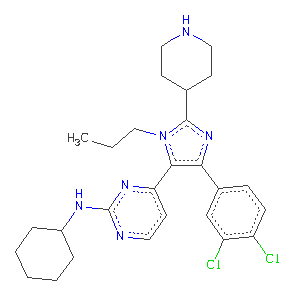
We'll start off with a model 1pmq.omit.pdb in which the waters and the ligand are removed, and refine this against the deposited structure factors 1pmq.mtz with water insertion using
refine -p 1pmq.omit.pdb -m 1pmq.mtz -L -d initial-omit | tee initial-omit.lis
to get an initial omit-map. This takes a while to run (about an hour on a 2006 PC),
so we've prepared files initial-omit-refined.pdb and initial-omit-refined.mtz. Load these into coot with
coot -p initial-omit-refined.pdb --auto initial-omit-refined.mtz
and have a look around. It's probably best to do Validate:Difference Map Peaks... ; the highest difference map peak corresponds to the chlorine in the para position
on the benzene ring. Notice that there is not so high a peak for the meta-chloro substituent.
Inserting the ligand
Get a CIF dictionary for compound 880 by running
grade_PDB_ligand 880
This will only work if you have access to mogul and OpenBabel; if you're doing this at a workshop these tools will be installed on the machine you're using; otherwise see the documentation link to be added. If you don't have access to these tools, download 880.grade_PDB_ligand.cif and 880.grade_PDB_ligand.pdb.
Load 880.grade_PDB_ligand.pdb into coot, and remove the hydrogens from it with the 'hydrogens in residue' option of the 'delete' icon, then move to the highest difference peak and
select Calculate:Move Molecule Here. Load the dictionary 880.grade_PDB_ligand.cif using File:Import CIF dictionary.
Now begins the frustrating task of convincing coot where the ligand goes. Turning off
the protein and the 2Fo-Fc density, using the Display Manager menu, probably helps;
use the Map button and ensure that you are fitting into Fo-Fc. Some people swear by
modifying chi angles, some people find that holding down CTRL and dragging a terminal
atom of a thoroughly misplaced group into the right place helps; however you prefer,
get the ligand into the density.
Now, choose the menu option Calculate:Merge Molecules... and insert 880.grade_PDB_ligand.pdb into initial-omit-refined.pdb; then save your work. Ensure that you save the merged initial-omit-refined.pdb rather than just the ligand position.
There's a really obvious water near N3, put that in (ensure you are adding it to the
right molecule using the chooser at the bottom of the 'insert water' dialogue) and save
again.
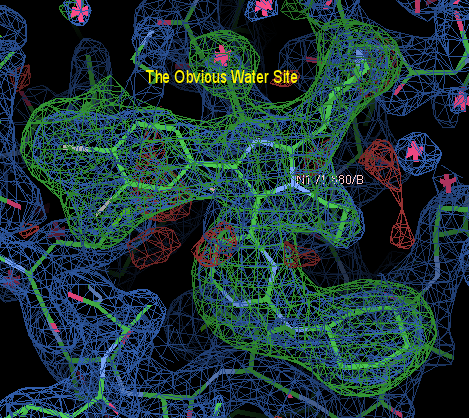
You can download the structure with the ligand inserted in a conformation that I like as inserted-ligand.pdb
A bit more refinement:
refine -p inserted-ligand.pdb -l 880.grade_PDB_ligand.cif -m 1pmq.mtz -d first-build-with-ligand -M ShortRunVoid | tee first-build-with-ligand.lis
Three things to notice here:
- This run does not use the -L option; there's no point in running that rather drastic automatic water insertion process more than once.
- We give BUSTER the high-quality dictionary using -l 880.grade_PDB_ligand.cif.
- We use the -M ShortRunVoid macro, which runs a short refinement suitable for regularising the structure and producing a good map. This macro should only be used on structures which have already seen BUSTER at least once, but is very much faster than a default refinement�.
Once again, you can skip ahead here by downloading the output first-build-with-ligand-refined.pdb and first-build-with-ligand-refined.mtz; you can download the refinement log first-build-with-ligand.lis which shows that the refinement took three and a half minutes.
Alternate ligand conformation and occupancy refinement
Water W9 (near residue A343) has strong positive difference density around it, indicating that it's something with more electrons than a water; the electron density looks somewhat tetrahedral, so let's make it a sulphate with non-unit occupancy ... add SO4 using the 'place atom at pointer' icon, and real-space refine it into place.
Then use the Measures:Residue Info menu option to
set the occupancy to something other than 1.0: the value of the occupancy is irrelevant,
it will be refined later, but the automatic occupancy refinement process only modifies
the occupancies of things whose starting occupancy is not 1.0.
Go to the ligand, residue 1 of chain B. There is negative difference density on the
CL45 atom, and positive difference density in the position opposite where it could
reside if you rotated around the C4-C35 bond.
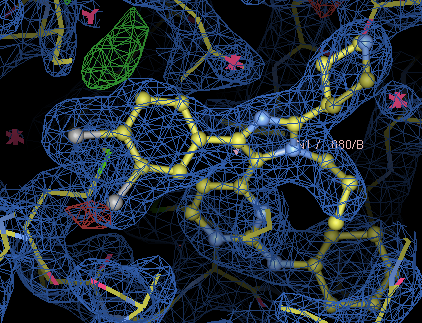
Ensure you've got 880.grade_PDB_ligand.cif imported, then add an alternate conformation to B1 using the 'add alternate conformation' icon.
Use the 'edit chi angles' icon to rotate the dichlorophenyl group.
Make any other modifications you like - should water W22 really be a water? how about
water W70? can you explain the density around proline A373 by a slight modification
to that bit of chain? could A111 have more than one sidechain conformation? then
save and do the re-refinement. Or collect tweak1.pdb if you don't want to do the fitting yourself.
This time we're going to refine occupancies.
Automatic occupancy refinement
Run
pdb2occ -p tweak1.pdb -o tweak1.gelly
to produce a gelly script describing which bits of the structure should be refined (see pdb2occ documentation), and then run
refine -p tweak1.pdb -Gelly tweak1.gelly \
-l 880.grade_PDB_ligand.cif -m 1pmq.mtz \
-d occref -nbig 3 | tee occref.lis
Note that we're not using the -M ShortRunVoid macro here; occupancy refinement converges relatively slowly and it is worth giving it three big cycles to settle. The run takes about fourteen minutes on our test machine.
While the refinement is running, you might want to load the tweak1.gelly file into a text
editor, open the gelly
manual in a web browser, and see if you can figure out what the commands in tweak1.gelly
actually do. Gelly has a powerful and flexible language for describing sets of atoms
and what to do with them, and pdb2occ can be used to produce examples of the use of the
gelly language to your heart's content.
The output is occref.pdb and occref.mtz: the alternative chlorine position (model in eye-searing pink) ends up at about 35% occupancy compared to the 65% of the tasteful green initial model, and the sulphate ends up at about 64% occupancy.
Looking at the final model in the last section, alternate positions are only really required for the dichlorophenyl ring. For most atoms of the ligand a single position is all that is required. In this case refinement with two complete alternates is OK but it entails doubling the number parameters used to describe the ligand. If the density is poor and noisy then two complete alternates can move apart to fit noise. So we could decide to model the ligand a single copy apart from dichlorophenyl ring that is the nine atoms CL45 CL46 C35 C37 C38 C39 C40 C41 C4 with two alternates. Compared to the previous approach this saves introducing extra parameters to describe the ligand. Every alternate atom has xyz coordinates and a B so a total of 26*4=100 parameters are saved.
The easiest (only?) way to make the changes is with an editor. Taking the result from the last stage I edited the ligand (residue 1 in the B chain):
- separating out alternate A and B for atoms CL45 CL46 C35 C37 C38 C39 C40 C41 C4
- changing the occupancies to be 0.5 for both A and B alternates of the nine atoms dichlorophenyl ring.
- deleting the B alternate for other ligand atoms
- removing alternate mark for the A alternate of other 26 ligand atoms. These atoms were given an occupancy of 1.0.
- the result of this editing is occref_edit_ligand.pdb
To refine this model:
pdb2occ -p occref_edit_ligand.pdb -o occref_edit_ligand.gelly
refine -p occref_edit_ligand.pdb -Gelly occref_edit_ligand.gelly -l 880.grade_PDB_ligand.cif -m 1pmq.mtz -d occref2 | tee occref2.lis
- once again the occupancy refinement default .Gelly file produced by pdb2occ works with no user-intervention.
- The refinement results in occref2.pdb and occref2.mtz
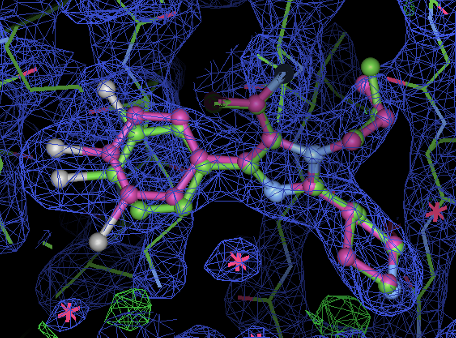
- The final occupancy for the A alternate in the ligand is 0.62 and the B is 0.37 (identical to previous).
- The ligand model looks like:
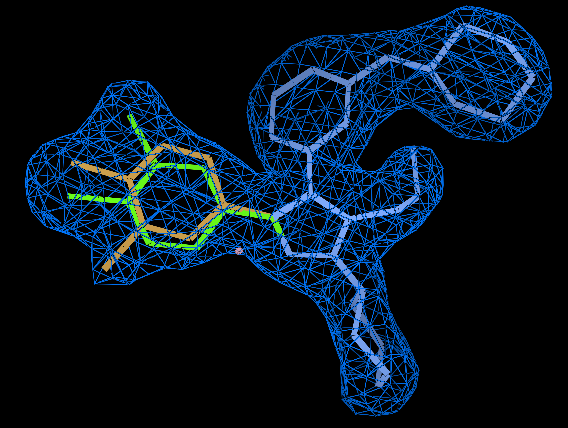
- Here the 2Fo-Fc has been masked except around the ligand and is contoured at 1.3 sigma. The ligand is colored so that atoms with no alternate and and occupancy of 1.0 are cyan. The A alternate ring (occupancy 0.62) is in green and the B (0.37) in orange.
Page by Tom Womack and Oliver Smart original version July 2010. Revised February 2011. Address problems, corrections and clarifications to buster-develop@globalphasing.com