Restraints play an important role in enabling the correct placement of ligands in protein-ligand complex structures as well as in their subsequent refinement (for in instance with BUSTER).
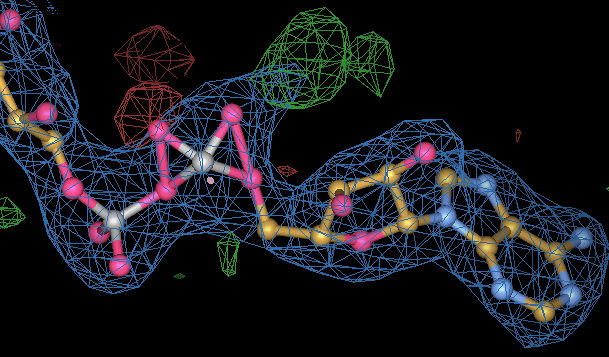 |
| If you do not take care and use good geometry restraints for ligands this is an example of what can happen. This is an FAD molecule from pdb entry 1yy5. The right hand phosphate group has been forced to umbrella by poor restaints. The poor geometry was noted in Kleywegt (2007) Crystallographic refinement of ligand complexes figure 3c, but remains in the current pdb. A simple BUSTER rerefinement of 1yy5 with a grade_PDB_ligand dictionary for FAD can fix the problem |
The BUSTER package has a number of approaches to produce restraints for ligands:
The restraint editor EditREFMAC is very useful for examining and editing REFMAC-style cif restraint dictionaries, such as those produced by grade.
See the appropriate section of the Buster FAQ for similar information about the handling of covalent ligands and LINKs.
Page by Oliver Smart, updated April 2013 and November 2020. Any questions regarding our software or this wiki should be directed to buster-develop@globalphasing.com