grade tutorial: different ways to use grade to produce cif dictionaries
- In this tutorial we are going to explore how grade can be used to create cif restraint dictionaries for dopamine.

- wikipedia dopamine article
- dopamine is small, so the examples should run quickly even on slow computers.
- The dopamine dictionary created can be in the final stages of the SULT1A3 tutorial example as this protein has dopamine bound in the active site.
- how grade is used depends on what your starting point is:
1 starting from SMILES
- Find the SMILES string for dopamine from the wikipedia dopamine article
- answer: c1cc(c(cc1CCN)O)O
- to create a dictionary with residue name LIG from the SMILES string either:
- Use grade on your workstation. You will need a CSD system licence so that you can run Mogul
- Note that it is important to wrap the SMILES in single quotes ' otherwise brackets get interpreted by the shell.
- use the grade command:
grade -resname LIG 'c1cc(c(cc1CCN)O)O'
- fill in the second page with the SMILES string and residue name as shown below
 click on image to view
click on image to view- the Grade Web Server will create a results page (normally in about 20 seconds). If server load is high then use the following link pre-calculated dopamine SMILES results page
- (Note that the Grade Web Server detects that the molecule supplied is dopamine. You should therefore use the existing 3 letter code and standard PDB atom names, see part 4.)
- running grade in either way will have created two files:
grade-LIG.cif grade-LIG.pdb
- the first is the cif restraint dictionary, the second some ideal coordinates in pdb format
- to check that the cif restraint dictionary is sensible do 2 things:
- start coot loading the dictionary by
coot --dict grade-LIG.cif
- in coot select "Extensions", "Modelling", "Monomer from dictionary" and type LIG.
- you should then see the dopamine in its ideal grade coordinates (these are contained in the cif dictionary).
- try doing a geometry regularisation by using the "Send in the bond angels" option this is the 4th item down in the left hand menu looking like a cross hairs with a red dot in it.
- start our cif dictionary editor with the command:
EditREFMAC grade-LIG.cif grade-LIG.pdb LIG
- this will fire up jmol and a table in which restraints can be viewed (and changed if wished).
- use this to note that grade by default creates lots of small 4 atoms planes because we think this is better allowing ring systems to gently bend.
- also note grade tries to create sensible torsion restraints for ligands. In this case an active N-C-C-ring 3 torsion with minimum of 180,-60,60 is created. This is based on the QM optimization because mogul supplies very few hits for this torsion.
- advanced exercise run mogul on the grade ideal coords, compare results to that for one of the dopamine from 2a3r.pdb: 2ar3_dopamine201.pdb
2. starting from a picture
- what if you do not have SMILES but simply a sketch.
- Use a sketcher to create SMILES.
- for instance: the Online SMILES Translator
- http://cactus.nci.nih.gov/translate/
- hit the Start Structure Editor button sketch your molecule and you see the smiles. You can check the SMILES is correct by using Daylight depict:
- http://www.daylight.com/daycgi/depict
- copy your SMILES there and see a lovely picture confirming that you are correct:
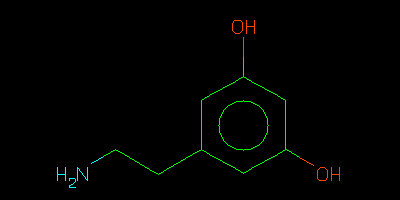
- Can you spot the error?
- I am sure you can do better. Once you have done so use the method in part 1 to produce a grade dictionary for dopamine.
3. Starting from ideal coordinates (with hydrogen atoms)
- Suppose you are supplied with the ligand described as an sdf file, often used in corporate databases
- L37831.sdf
- simply use this by
grade -in L37831.sdf -ocif L37831.cif -opdb L37831.pdb
- Use EditREFMAC to examine the dictionary, (see part 1). Can you recognise the molecule? It is likely that you see density for glycerol sooner or later!
- The Grade Web Server http://grade.globalphasing.org/ is currently restricted to taking ideal coordinates in mol2 format only, with all hydrogen atoms and with atoms having unique names only. (This will soon be improved!)
4. Producing cif dictionary for an existing PDB ligand
- Suppose you want to use a grade dictionary to regularize the dopamine from 2a3r pdb entry 2ar3_dopamine201.pdb in coot.
- Existing pdb ligands have defined 3-letter residue codes and atom names (including for the hydrogen atoms). Search for dopamine at either:
- rcsb ligand expo http://ligand-expo.rcsb.org/
- or pdbechem http://pdbe.org/pdbechem
- What is its 3-letter code?
- You could download ideal coordinates in pdb format, convert to our preferred mol2 format with openbabel and then use grade by hand.
- but this is a pain so
grade_PDB_ligand LDP
coot -p 2ar3_dopamine201.pdb --auto LDP.grade_PDB_ligand.cif
- The dopamine dictionary created can be in the final stages of the SULT1A3 tutorial example as this protein has dopamine bound in the active site.
Page by Oliver Smart 08 Feb 2011, updated for for Grade Web Server 09 March 2012. Any questions regarding our software or this wiki should be directed to buster-develop@globalphasing.com
 click on image to view
click on image to view