FAQ: rhofit has found the correct site to fit but the ligand has not been fitted correctly
Things to check:
- is the cluster file used in the fit sensible? Does it cover all the difference density? These easiest way to check this is to use the visualise-rhofit-coot visualiser and click on the "Cluster visible" tick box. If the cluster does not cover all the difference density then it might be worthwhile manually producing cluster files.
- could the bad fit be caused by a poor ligand dictionary?
- rhofit uses refinement restraints to work out how it can adjust a ligand's conformation to fit the density.
- The misidentification of planes is probably the biggest problem in restraint dictionary generation. Plane problems are often behind rhofit failures.
- To quickly visualize what plane restraints are being used for the ligand in rhofit, you can use the EditTNT program
- simply cd to rhofit results directory and issue the command:
EditTNT best.dic best.pdb
- for instance, using rhofit to fit the "880" ligand to 1pmq using libcheck to generate a cif dictionary from SMILES results in a poor fit.
- EditTNT shows that the three unsaturated rings are held in a single plane so cannot rotate relative to one another.
 (click on image to zoom in)
(click on image to zoom in)- the density clearly shows this is incorrect.
- If the input cif dictionary is edited splitting the plane into 3 separate planes the fit works.
- If it still doesn't work, please let us know, giving all the details you're allowed of the input PDB, MTZ file, and ligand.
Page by Oliver Smart original version 1 April 2010. Address problems, corrections and clarifications to buster-develop@globalphasing.com
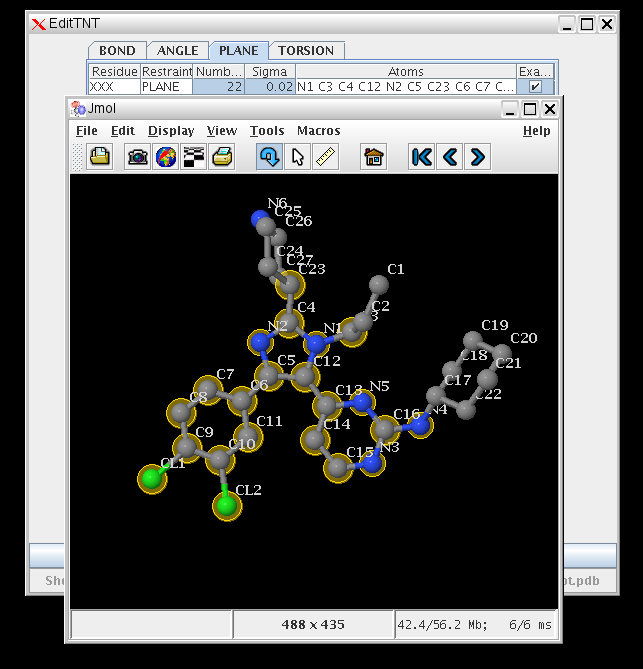 (click on image to zoom in)
(click on image to zoom in)