Pipedream Tutorial 5
(revision 1.0, February 2015)
| Note | The features described in this tutorial require Pipedream version 1.1.0 or later. |
|---|
This example illustrates the use of Pipedream with multiple input models, in this case using a number of beta-secretase structures. The experimental data is taken from PDB id: 4KE1. The three input models used are PDB id's: 1W50, 4DH6 and 4J0P.
| 4ke1.mtz | the structure factors as supplied by the pdb for 4ke1, converted to mtz format |
| 1w50.pdb | the model coordinates as supplied by the pdb for 1w50 |
| 4dh6.pdb | the model coordinates as supplied by the pdb for 4dh6 |
| 4j0p.pdb | the model coordinates as supplied by the pdb for 4j0p |
- (A) Introduction
- (B) Input model preparation
- (C) Identification of conformational change
- (D) Restraint dictionary generation
- (E) Pipedream run
- (F) Analysis
Conformational change in proteins is a well-known and studied phenomenon. Such changes can be extremely localised, limited to alterations in side-chain conformation, or they can be much much more extensive, such as rigid body domain movements. Localised loop movements are frequently observed in proteins, particularly in response to ligand / cofactor binding.
Such loop movements can be of a large enough magnitude that refinement alone is unable to deal with them - hence it is important to pick the correct input model for refinement. For example, if you are looking at a protein where a loop occludes the known ligand binding site in its apo state, but moves out of the site in response to the presence of a ligand, then it does not make sense to use an apo model, with the loop in the in conformation, in refine ment against data where a ligand is bound. Refinement alone is unlikely to move the loop out of the binding site and subsequent ligand fitting will fail, resulting in a false negative. Conversely, it is similarly unwise to use as input to refinement a model for a conformation of the protein in a case where a ligand is bound if the experimental structure is in the apo conformation, this time resulting in a false positive.
However, in the context of running a fragment/ligand screening pathway, how do you know ahead of time whether or not the soaked ligand has bound and therefore which input model to use with Pipedream?
Pipedream deals with this issue by allowing the input of multiple models, performing an initial refinement on all of them, after which it makes a decision as to which best matches the experimental data. It will then carry on with refinement and ligand fitting using this model alone.
The method employed currently is designed to distinguish between conformational changes produced by main-chain differences, such as loop movements. Additional, limited domain movement (such as hinge-bending) can also be accommodated by running Pipedream with an appropriately constituted rigid body definition file, so that the initial refinement can correct for relative domain shifts before model comparison.
However, the current method is not sensitive to conformational change caused solely by side-chain movements.
As described in the program documentation, any ligands or other compounds present in the input models, that are not expected in the experimental data must be removed from them.
In this case, 1w50 is a native model and is fine. However, both 4dh6 and 4j0p contain ligand coordinates (0KN for 4dh6 and 1H8 and DMS for 4j0p) and these should be removed from the pdb files. Note that any LINK cards referencing removed coordinates must also be removed.
egrep -v " 0KN " 4dh6.pdb > 4dh6-input.pdb egrep -v " 1H8 | DMS " 4j0p.pdb > 4j0p-input.pdb
Strict requirements of Pipedream where multiple input models are input are that as well as all having the same space group, cell dimensions and sequence to the experimental data, a) all of the input models must be superimposable and b) they must all share a common residue numbering and chain identification scheme.
Now we must ensure that all 3 of the input models are superimposable.
We can use CCP4 program gesamt to do this:
gesamt 4dh6-input.pdb 1w50.pdb > 4dh6.pdb gesamt 4j0p-input.pdb 1w50.pdb 4j0p.pdb
1w50 and 4dh6 both share a similar residue numbering. However, although 4j0p has the same sequence to 1w50 and 4dh6, the residue numbering in the PDB file does not match. This MUST be corrected before it can be used in Pipedream.
The first residue in both 1w50 and 4dh6 has residue number -1. However, the equivalent residue in 4j0p has residue number 60 (an offset of -61).
Open the superimposed 4j0p coordinates in coot and renumber, applying an offset of -61. Save the modified coordinates.
This should give you three input models that are superimposable and share a common residue numbering scheme.
(C) Identification of conformational change
Where multiple models are input, Pipedream needs to determine the regions of the structure that are conformationally different. It can do this in two ways.
The preferred method is for the user to pre-determine the important regions of the protein where there are conformational differences between the input models and supply this information to Pipedream.
Alternatively, if left undefined, Pipedream will attempt to identify these regions automatically by analysis of pairwise RMS deviations of the superimposed models - see the pipedream documentation for details.
Pre-determination of the conformational changes is preferred as it allows more specific targeting of changes that are close to the expected ligand binding site or are structurally significant. The automated method will consider differences throughout the entire structure.
Use coot to analyse the superimposed structures to identify significant differences.
Overall, the three models are extremely similar. However, there is a marked difference in position of a loop (res 68 - 75) over the protein active site, as shown below:

We need to create an input file listing the residues that are conformationally different. As the documentation describes, this file needs to contain a comma separated list of these residues, in the form of <res name> <chain id> < res number> - see seq.list.
(D) Restraint dictionary generation
4KE1 was complexed with a large and flexible inhibitor, 1R6.
- We need to generate a restraint dictionary for 1R6, using grade2:
grade2 -P 1R6
We are now ready to run Pipedream.
We will do this twice. Firstly lets run Pipedream using the preferred method of defining the region of conformational change:
pipedream -hklin 4ke1/4ke1.mtz -nofreeref -xyzin 1w50.pdb,4dh6.pdb,4j0p.pdb \ -rhofit 1R6.restraints.cif -rhothorough -seqin1 seq.list -d seqin
- The -seqin1 option is used to specify the file containing the list of residues to analyse for differences.
- The ligand (1R6) is quite large and flexible, so we have specified the -rhothorough flag, to tell Rhofit to run more trials than usual.
For the second run, we will repeat the above command, but without -seqin1 seq.list. Here, Pipedream will determine where there is conformational change automatically.
pipedream -hklin 4ke1/4ke1.mtz -nofreeref -xyzin 1w50.pdb,4dh6.pdb,4j0p.pdb \ -rhofit 1R6.restraints.cif -rhothorough -d auto
The main output file from the first run, summary.out, indicates that the job has run correctly:
- the limited MR section of the file shows that very good MR solutions were found for all 3 input models
- the model selection panel shows that Pipedream has picked out a very clear solution - 4dh6 has a significantly higher mean Z-score over the residues identified in seq.list (9.1) than either 1w50 (5.5) or 4j0p (5.1).
- Pipedream identified 4dh6 as the model that best matches the input data and all subsequent steps carried on using only 4dh6.
- Rhofit has found a single solution for the ligand, with a good score and post-refinement has completed nicely.
But is the outcome correct?
The final R/Rfree after post-refinement from this run of Pipedream are 19.5 / 23.3.
These compare very favourably with the quoted R/Rfree of 21.5 / 24.6 for the deposited 4KE1 structure (although this was refined with Refmac).
The picture below shows the ligand in the final post-refined structure. The actual deposited 4KE1 model is also shown, in green.
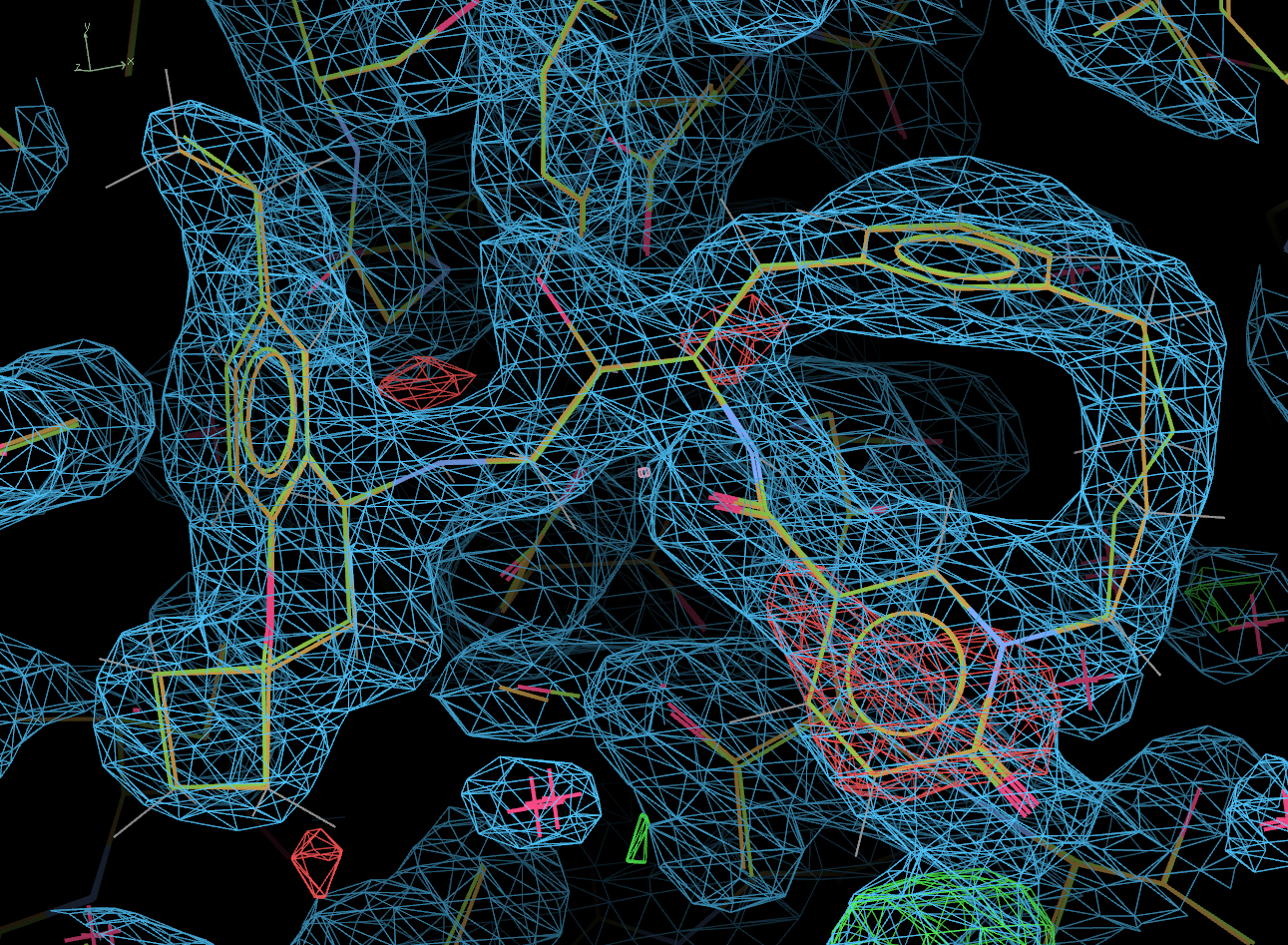
Clearly, other than a minor difference at one position, the ligand fit is correct.
What about the loop that was identified as different in the 3 input models. The picture below shows this loop, above the ligand. Again, the 4KE1 model is shown in green.
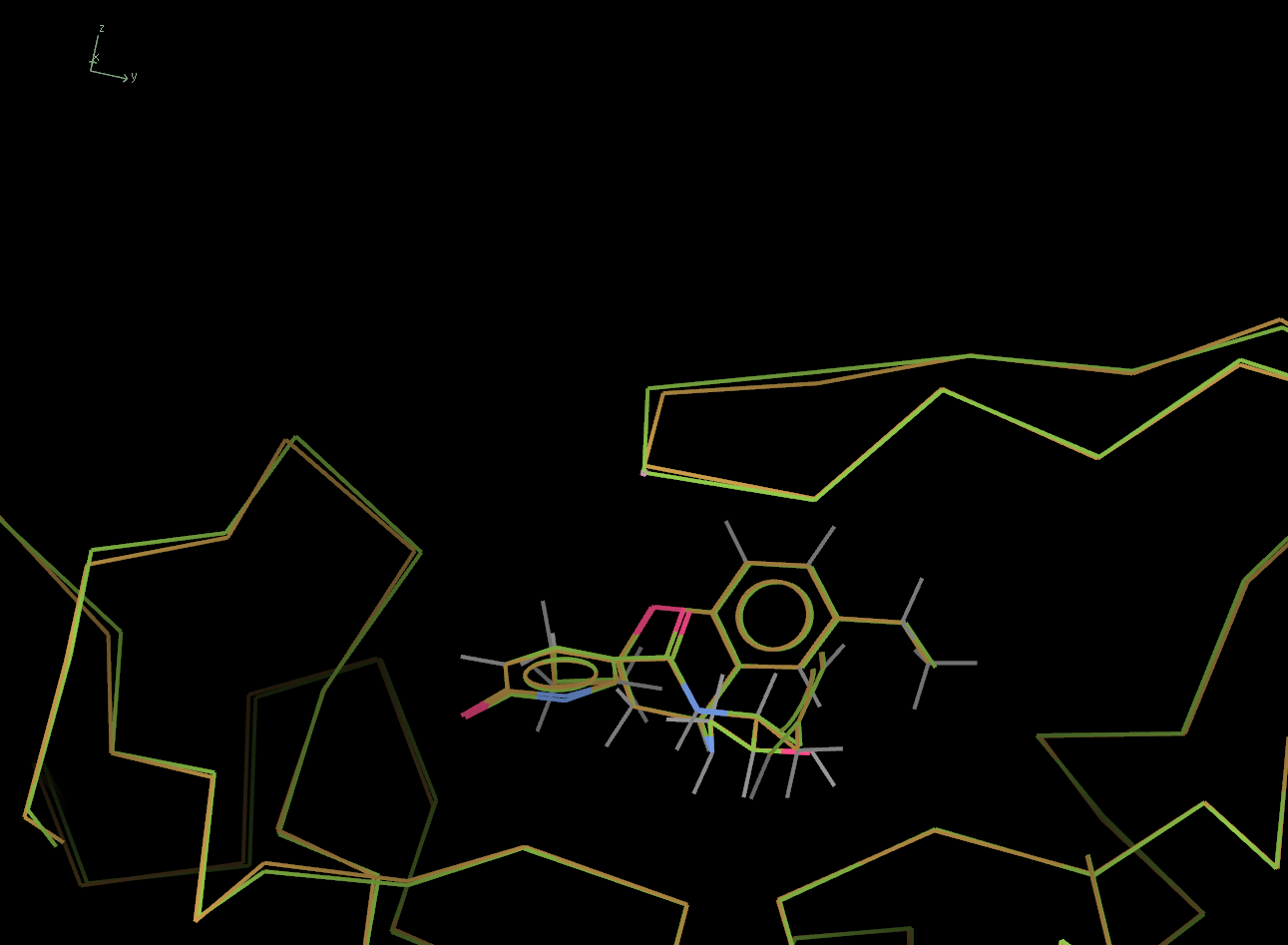
Now lets look sat the output from the second run of Pipedream, run with automatic conformational change detection - see summary.out
- The outcome is essentially the same as with the above run.
- The correct solution (4dh6) was also clearly identified, although the difference in mean Z-score is not quite as pronounced.
- The residues selected by the automatic procedure are here. You will notice that the main loop has been identified but two additional residues, both remote from the loop and from each other, have also been selected.
- The final R/Rfree after post-refinement and the final model are very similar to the first Pipedream run.
So, both Pipedream runs, run where the region of conformational difference between the models has been defined manually and when determined automatically, have successfully identified the input model that best fits the experimental data and have correctly fit and refined the bound ligand.