This is an attempt to simulate a ligand-fitting pipeline.
The MEK1 kinase is used as the example protein; there are 13 hits on 'MEK1' in the PDB, of which eight were considered:
| Structure | Ligand bound | Molecular-replacement model used |
| 3e8n | VRA | 2p55+ATP+MG |
| 3dv3 | MEK | 3e8n+ATP+MG |
| 3dy7 | 1CX | 3e8n+ATP+MG |
| 3eqc | 3BM | 3eqd+AGS+MG+CA+NA |
| 3eqf | KSA | 3eqd (KSA binds at the AGS site) |
| 3eqg | 4BM | 3eqd+AGS+MG+CA+NA |
| 2p55 | MRA | 3e8n+ATP+MG |
| 1s9j | BBM | 3e8n+ATP+MG |
For each structure, the EBI SSM tool was used to find a structure with high sequence similarity and good RMS distance; this was usually another one of the eight. Waters were removed, though (since these are all cases in which the ligand binds next to the ATP side) the ATP (or ATP analog) and bound magnesium were left in, and the model was placed into the deposited structure factors by molecular replacement.
After molecular replacement, the structure was refined by a three-step procedure
refine -p molrep.pdb -m data.mtz -autoncs -d FIRST FitMAP -p FIRST/refine.pdb -m FIRST/2Fo-Fc.map -dm FIRST/Fo-Fc.map refine -p protein_fitted.pdb -m data.mtz -autoncs -L -d SECOND
where the FitMAP run adjusts side chains, since there were several cases where side-chains near the ligand position had moved.
Ligand models were prepared using an internal tool (under development) which takes a very simple text description of the ligand topology (flat rings, tetrahedral centres, single and double bonds and chemical element), adds hydrogens where atom valencies are not fully filled, 'inflates' it to a 3D model with all angles constrained to either 120 or 109.5 degrees and all bond lengths equal to modal values for bonds between atoms of the appropriate type, and uses the AM1 quantum chemistry tool to find a minimum-energy conformation starting from that 3D model. Hydrogen atoms are then removed since the dictionary-generation tool used by rhofit does not yet handle them adequately.
rhofit was then run
rhofit -lp ligand.pdb -p SECOND/refine.pdb -m SECOND/refine.mtz
the best rhofit hit was added to the model in SECOND/refine.pdb and a third refinement performed:
refine -p merged.pdb -m data.mtz -autoncs -d THIRD
The correlation coefficient of the refined ligand with the density was then computed, and the result compared with the deposited structure for the ligand.
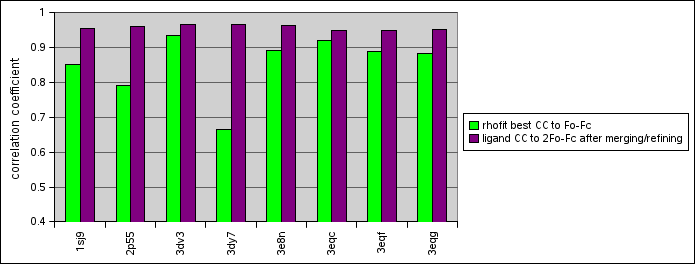
(correlations in the first column are from rhofit and to the Fo-Fc from the refinement; the second column of correlations is against 2Fo-Fc for a model refined with the ligand in place so should be expected to be higher)
| Structure | CC for Hit_00_000.pdb | Correlation after merging in and refining | Comments on fit compared to PDB answer |
| 1s9j | 0.8521 | 0.9547 | Identical except for twist of flexible tail |
| 2p55 | 0.7910 | 0.9587 | Rings in right place, but there's a water placed where the acetylene group in the ligand should go |
| 3dv3 | 0.9337 | 0.9652 | Rings perfect; flexible tail goes in wrong direction because of water insertion near cluster |
| 3dy7 | 0.6647 | 0.9646 | Identical except that the tail without density points the other way in our solution |
| 3e8n | 0.8898 | 0.9619 | Pretty much perfect; and not misled by a patch of unbuilt main-chain nearby |
| 3eqc | 0.9200 | 0.9497 | Rings perfect, iodine atom somewhat anisotropic, some difference density near oxygen of tail |
| 3eqf | 0.8887 | 0.9477 | Pretty much exactly right |
| 3eqg | 0.8819 | 0.9501 | Rings perfect, oxygen at end of wiggly part points in wrong direction |
 data presented as bar chart.
data presented as bar chart.
Page by Tom Womack. Original version 2 November 2009. Address problems, corrections and clarifications to buster-develop@globalphasing.com