Content:
Introduction
For this protein we know:
autoSHARP
You should have a reflection file from the autoPROC tutorial - if not: just use 1o22_autoPROC.sca. You can upload this file (as well as the above sequence file) through a link on the SHARP Control Panel:
Starting the job
You should "Start" autoSHARP based on "None" and then describe the type of experiment:
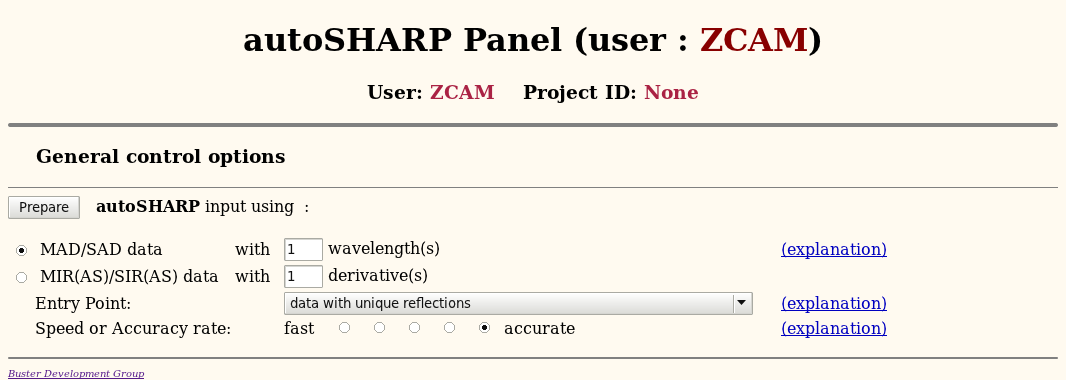
Just leave everything at their default value and hit "Prepare". This will get you to the main autoSHARP input panel - where you will see several sections:
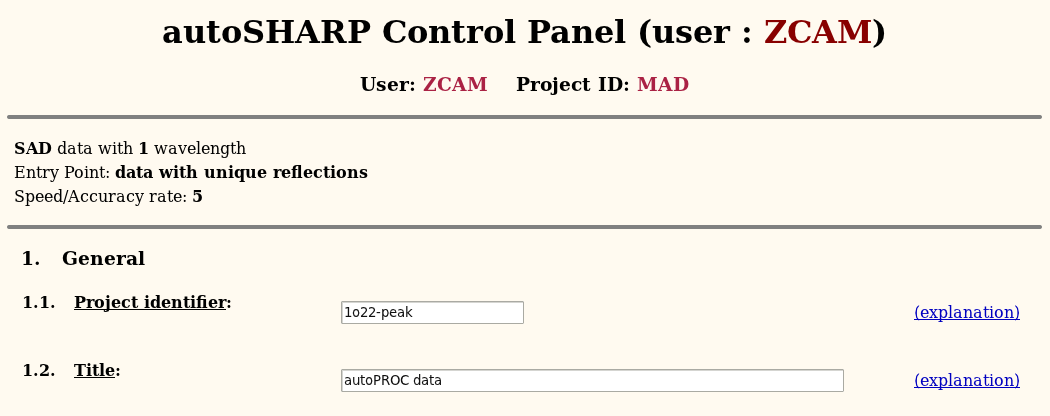
You want to give a fairly descriptive identifier (without special characters, spaces or other funny characters - since directories and files will be created using this string).
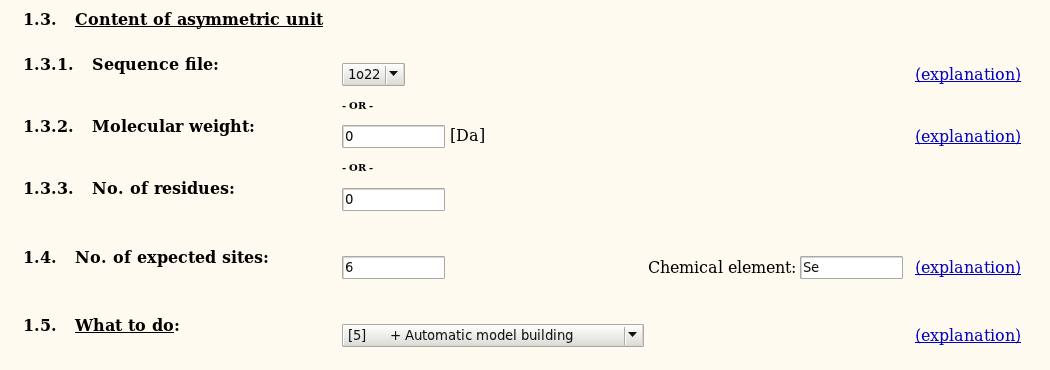
Select the sequence file (containing the monomer sequence) and give the number of expected heavy atoms (per monomer) as well as the atom type. Usually, autoSHARP can be run all the way to automatic model building - but you could also restrict it only up to density modification.
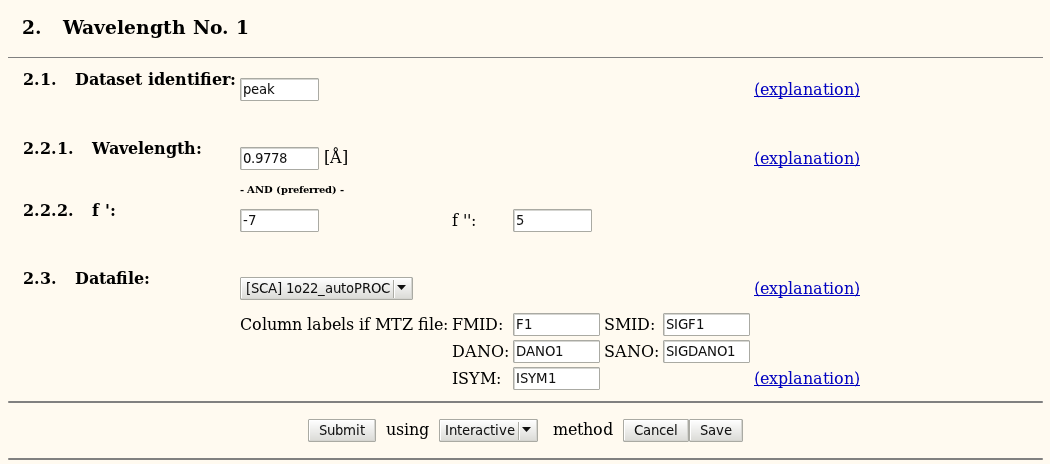
For each dataset, some information is required: wavelength and f'/f'' values. If using *.sca files, the fields for column labels can be ignored.
After that, all that is required is to hit "Submit".
Interpreting output
While looking at the main autoSHARP output file, some points are of interest:
- that file is being written to while you are viewing it - so from time to time hit the "reload" button of your browser (or Ctrl-R on the keyboard).
- the main output file is rather terse - but each point will contain a link to "(details)" to see more information about this particular step. Furthermore, each step might again contain further links to even more detailed logfiles. you might sometimes need to go back one or more levels to reach again the main autoSHARP logfile. Or use the "results" browser from the main "SHARP Control Panel"
- an explanation link should bring up the relevant part of the online documentation
The first stage gives some general information - mainly of interest to ensure all files are read correctly:

But it also contains an automatic analysis of the most likely number of molecules per asymmetric unit. Further analysis is mostly interesting for cases with several datasets - so slightly boring here:

It might be worth checking the self-rotation analysis if one expects more than 1 mol/asu.
The next step is heavy-atom detection with SHELXD:
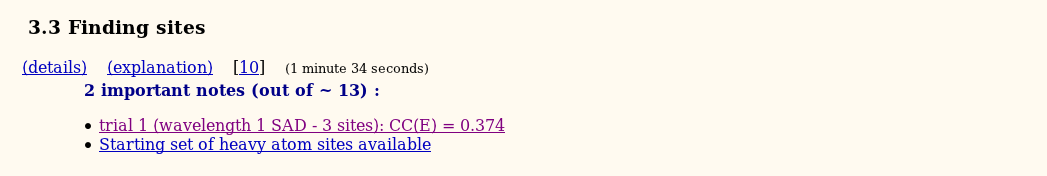
Several graphical plots are provided to check for success and quality of the substructure solution.
The first run with SHARP (using the set of initial sites) should already give some meaningful phase information:
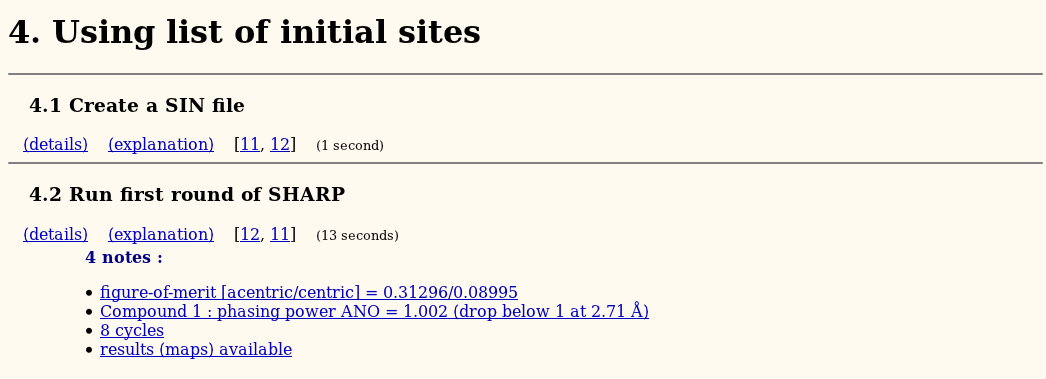
Automatic interpretation of LLG residual maps might allow placing further (weak) heavy atoms, which could improve the phasing further. this procedure is iterated until convergence:

At this point two phase sets are available: the original hand (in the original spacegroup) and the inverted hand (in it's enantiomorph spacegroup ... which could be identical).
Now we need to decide which is the correct hand/enantiomorph:

After density modification in the most likely hand/spacegroup, a map should be available - which hopefully is good enough to see secondary structure. This is then passed on to automatic model building (if requested) or you could use it outside autoSHARP in another program.
Did it work? If after about 30 minutes (on a i7-2720QM laptop) you get a structure like this
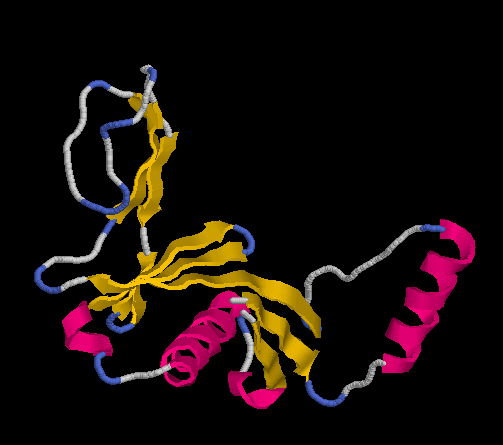
it did.