Grade2 Changelog¶
Please check the online version of this issues page: https://gphl.gitlab.io/grade2_docs/changelog.html for updates.
v 1.5.0¶
- Distributed with
BUSTER 202401??
- Build Date
12 January 2024
New Features¶
Dative bonds in 2D schematic diagrams are now marked with dotted lines rather than arrows. For instance for heme
HEM: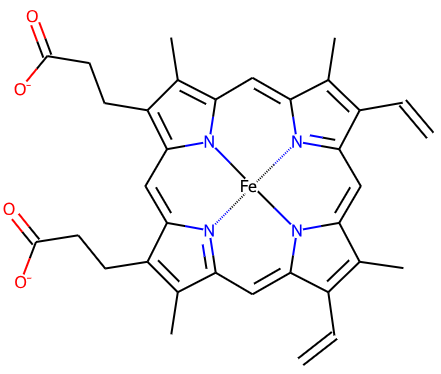
Previously arrows were used (see below) but dotted lines are more standard and clearer. (#633)
Performed an analysis comparing Grade2 and EH99 Restraints for amino acid side chains and introduced a new option --eh99_sigma_correction that scales up all non-hydrogen bond and angle sigmas to match the mean sigma values of the EH99 amino restraints used by BUSTER. (#635)
Fixes¶
Fixed a major bug where a plane was defined for torsions that the MMFF94s force field held flat, when there was an insufficient number of Mogul hits to assess planarity. This leads to incorrect plane assignment, particularly for bonds between two aromatic rings. Now the MMFF94s 2-fold torsion term is only used to assign planarity for hydrogen atoms.
Thanks to Markus Rudolph for reporting this bug. (#636)
The --big_planes option now orders atoms more sensibly using numbers in the atom ID rather than alphabetically. This is intended to make any editing of the big plane easier. Please note that the --big_planes option overemphasizes ring planarity and should be used with caution.
Thanks to Markus Rudolph for reporting this improvement. (#637)
Fixed bug where using the --antecedent option with a restraint dictionary for a related molecule that matches only part of a large complex ligand can result in Grade2 terminating with error message containing
atom index out of range.The problem is caused by difficulties in creating 2D coordinates with the matching atoms being aligned. If the problem occurs then Grade2 will now include the following warning message in its output:
WARNING: - ANTECEDENT: error in computing aligned 2D coordinates, so the WARNING: - ANTECEDENT: 2D diagrams and coordinates will not be aligned. WARNING:
Grade2 will then proceed and produce 2D coordinates and 2D schematic diagrams without alignment. (#632)
Checked that Grade2 can handle five-character PDB chemical component IDs. The PDB will soon be issuing five-character PDB chemical component IDs as three-character IDs are exhausted. Grade2 has no problem in handling five-character chemical component IDs. But programs that use PDB-format coordinate files (such as BUSTER) will have problem with chemical components IDs longer than 3-characters. One possible solution is to use the --resname option to set a working ID such as
01for the chemical component, in place of the five-character PDB chemical component ID. (#640)
Fixed bug where Grade2 erroneously imposed a planar restraint on 3-atom rings. This makes no sense as 3 points define a plane, and so BUSTER terminates with an error if supplied with a 3-atom plane.
Thanks to Clemens Vonrhein for raising this issue. (#641)
Fixed bug where Grade2 did not properly handle PDB ligands with commas in atom IDs (for example T46).
Thanks to Clemens Vonrhein for raising this issue. (#643)
Fixed bug where Grade2 crashed for malformed PDB chemical components: the original releases of VLW and VMI that both had a nitrogen atom wrongly assigned a charge +4. Grade2 can now handle the problematic definitions and we will supply the PDB with information to correct both the ligands.
Thanks to Clemens Vonrhein for raising this issue. (#646)
The --PDB_ligand --rcsb option will now first download information from a URL like https://files.rcsb.org/ligands/download/ABC.cif , as this is now the approved URL to obtained for this information.
Thanks to the RCSB for the update. (#657)
v 1.4.1¶
- Distributed with
BUSTER 20230726
- Date
18 July 2023
This release contains two fixes.
Fixes¶
The CSD update 2023.2 (July 2023) introduces a change that means that old Grade2 versions prior to this release
1.4.1will not work with it. This release will work with both the CSD update 2023.2 and with older versions of the CSD. Please see the CSD compatibility chapter https://gphl.gitlab.io/grade2_docs/csd_compatibility.html for further details.
Fixed bug where Grade2 produces restraints with bond angle sigma set to zero that causes BUSTER to terminate. Now a minimum value of 0.3º is used for bond angle sigmas (CIF item
_chem_comp_angle.value_angle_esd). A similar protection is now also applied to bond restraints with a minimum value of 0.005Å for the bond sigma (CIF item_chem_comp_bond.value_dist_esd). The minimum values were set by surveying the range of sigmas in the set of restraints for common compounds distributed with BUSTER.Thanks to Christian Schleberger for reporting this bug. (#622)
v 1.4.0¶
- Distributed with
BUSTER 20230614
- Date
22 May 2023
New Features¶
The 2D coordinates used for schematic 2D molecular diagrams are now also stored in the output CIF-format restraint dictionary. The CIF category
pdbe_chem_comp_atom_depictionis used for the records (as this is used in PDBe CCD files). Storing the 2D coordinates will facilitate the production of consistent 2D diagrams in a future validation tool. (#549)
The schematic 2D molecular diagrams SVG output files are now scaled to be clear for both small and large ligands. Previously the same sized diagram was used regardless of ligand size. (#569)
Molecules where RDKit needs to add a hydrogen atom to show the chirality in the schematic 2D molecular diagrams (such as
MOIandTXL) are now shown without any chiral wedges as this produces clearer pictures. (#572)
The font size for atom labels used for 2D diagrams has been slightly reduced. Chiral wedges are no longer shown in diagrams with atom labels. This makes the diagrams clearer. (#573)
For PDB chemical components the 2D coordinates used in schematic 2D molecular diagrams are now taken from PDBe Data-enriched chemical component definitions (PDBe CCD), where possible. Please note if the command line option --rcsb is used then the default wwPDB CCD will be used and as this lacks the data-enrichment records RDKit generated 2D coordinates will be produced. (#142)
The PDBe CCD 2D depiction coordinates are produced by the
ccdutilstool https://github.com/PDBeurope/ccdutils . In many cases they are superior to the default RDKit depictions. For instance, for compounds containing porphyrin rings, such asHEM, Grade2 now creates a clear schematic 2D molecular diagram: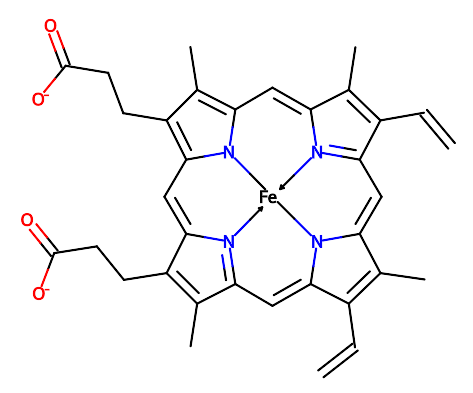
Here the arrows are used to show dative bonds to the iron atom.
To see how the use of PDBe CCD coordinates combined with other improvements to the 2D coordinates produce better results, contrast the 2D diagram for the PDB component
MOImorphine with atom labels, produced by this release with the cluttered diagram produced by the previous releases: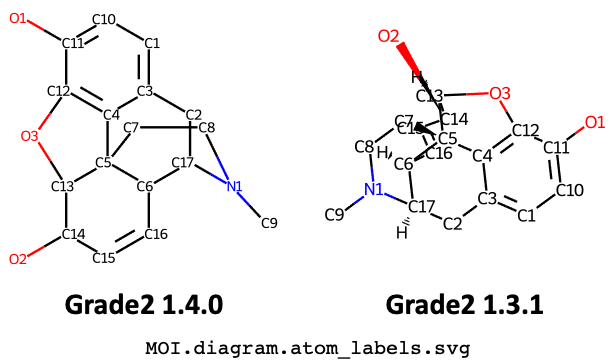
New options --antecedent RELATED_RESTRAINTS_CIF and --antecedent_disregard_element RELATED_RESTRAINTS_CIF that base the atom IDs and 2D coordinates for schematic 2D diagrams on the Grade2 restraint dictionary of a related ligand are introduced. If the input has atom IDs then these are disregarded and new atom IDs are assigned. For more information see the Atom Naming chapter.
Thanks to Steven Sheriff for suggesting this extension. (#548 #550 #577)
A new option -R, --rdkit_canonical_atom_ids sets new atom IDs based on the atom order in the RDKit canonical SMILES string for the molecule. As the canonical SMILES string is independent of order of the atoms in the input molecule, the same atom IDs will be produced regardless of the source. The atom IDs produced
--rdkit_canonical_atom_idsare straightforward with adjacent atoms generally having adjacent IDs, where possible. For more information, please see the Atom Naming chapter.Please note, that using
--rdkit_canonical_atom_idswipes out any existing atom IDs and reorders the atom list. If you would like Grade2 to write out an alias table showing the mapping between the original and new atom IDs, please let us know. (#592)
A new option --inchi_canonical_atom_ids sets new atom IDs based on the canonical atom order created when calculating the InChI of the molecule. The procedure is based on the ALATIS Unique Identifiers proposed by Dashti et al. (2017) and non-hydrogen atom IDs should be identical to ALATIS. It should be noted that``--inchi_canonical_atom_ids`` adjacent atom IDs are normally far apart in a molecule, making them less "user-friendly" but more universal than
--rdkit_canonical_atom_ids. For more information, please see the Atom Naming chapter.Please note, that using
--rdkit_canonical_atom_idswipes out any existing atom IDs and reorders the atom list. (#132)
CSD-based restraints for pentafluorosulfanyl SF5, that keep the group in an octahedral geometry, have been introduced. Please see the Difficult Chemical Groups chapter for a full description.
Thanks to Michael Blaesse for suggesting this improvement. (#518)
A new option --chiral_non_carbon will add chiral restraints specifying the configuration for all chiral atoms not just tetrahedral carbon atoms; so nitrogen, sulfur, and phosphorous atoms will be treated as defined chiral centers.
The command line arguments used in a Grade2 run are now recorded in the restraints CIF as item
_gphl_chem_comp_info.arguments. Similarly item_gphl_chem_comp_info.run_daterecords the date of the run. (#552)
Fixes¶
Fixed Grade2 to work following the large alterations to the CSD directory structure made in the April 2023 CSD release. The fix allows Grade2 should to work both with the new CSD release as well as with previous CSD releases, provided the environment variable
BDG_TOOL_MOGULto is set to the location the Mogul executable on your system as explained in the Installation Chapter. (#580)
The default --PDB_ligand option will now first download information from a URL like https://www.ebi.ac.uk/pdbe/static/files/pdbechem_v2/ATP.cif using the https protol before falling back to ftp. This has the advantage that the https protocol is used and consequently is unlikely to cause firewall connection issues. (#587)
When RDKit cannot calculate atomic partial charges no longer give a
WARNINGmessage. Partial charges are not commonly used by crystallographers and so the message is unnecessary. Any user of the restraint dictionary who wants partial charges can easily notice they are all zero.Thanks to Clemens Vonrhein for suggesting this improvement. (#589)
When adding a proton to groups likely to be charged at neutral pH during the Charging process five character atom IDs are now avoided to allow a PDB file to be written.
Thanks to Clemens Vonrhein for suggesting this improvement. (#593)
v 1.3.2¶
- Distributed with
Grade2 Update Release 08 March 2022
- Date
06 March 2023
This release contains one backwards compatible bug fix.
Fixes¶
Fixed bug where Grade2 wrongly warns and assigns ambiguous chiral restraints for SDF input files produced by some programs.
The problem is that SD files from different programs vary in the markup of whether the chirality of the molecule is set. The problem is that SD files have multiple places where the chiral configuration can be specified:
The 3D coordinates (if present)
The COUNTS line chiral flag
On individual atoms in the ATOMS block (often written but seldom read?).
On individual bonds in the BONDS block
SD files produced by different programs will often not set some or all of these, Please see https://depth-first.com/articles/2021/12/29/stereochemistry-and-the-v2000-molfile-format/ for a discussion of the problem. This bug is an unintended consequence of bug fix #559 in Grade2 release
1.3.1which meant that instead of simply basing chirality on the 3D coordinates supplied within the SD file it also required that appropriate chiral flags be set with the BONDS block.The bug fix alters behaviour back to that of previous Grade2 releases where the chirality of the molecule is taken from the 3D coordinates (just like for MOL2 files). To make the source of chiral restraints clear the terminal output now includes a line:
Note that chiral restraint configuration is set from the 3D coordinates
Thanks to Dirk Reinert for reporting this bug. (#570)
v 1.3.1¶
- Distributed with
BUSTER 20230217
- Date
05 February 2023
This release contains only backwards compatible bug fixes.
Fixes¶
Grade2 will now write all CIF categories as loops, even if they only contain a single item. An exception is made for category
gphl_chem_comp_infowhich by default is written using key-value pairs as this makes inspection easier. If you want all CIF categories includinggphl_chem_comp_infoto be output as loops then set the environment variable BDG_GRADE2_CIF_LOOP_ALL to"yes".The change will help other programs that are restricted to read restraints in CIF loops (rather than fully supporting the CIF standard). For example, Coot
0.9.6that does not read a single bond restraint or a bond angle restraint written as a key-value CIF category. Grade2 restraint dictionaries with a single bond or bond angle restraint can now be read in.Thanks to Steven Sheriff for suggesting this alteration. (#555)
Fixed bug found for Grade2 with an input SMILES string having some chiral center configurations specified but some left ambiguous. Grade2 now produces an output restraint dictionary where the chiral restraint volume is set to
bothfor the ambiguous centers rather than being arbitrarily assigned. A warning message is now written when there are any ambiguous chiral centers.Thanks to Meigang Gu for reporting this bug. (#559)
Fixed bug where Grade2 terminates with
IndexError: string index out of rangefor large ligands that have more than 26 five-membered or six-membered rings.Thanks to Deepak Deepak for reporting this bug. (#532)
Fixed bug where Grade2 produced SHELXL restraint files with
REMcomment lines longer than 80 characters.Thanks to Tim Gruene for reporting this bug, using the Grade Web Server. (#544)
Fixed bug where a mangled MOL2 file with valency problems causes a Grade2 crash on checking amino acid labelling.
Thanks to a user of the Grade Web Server from China for raising this problem. (#545)
Fixed bug where Grade2 crashed when using --pubchem_names with a ligand that PubChem cannot standardize, like a peroxide ion [O-][O-]. (#538)
Fixed bug where Grade2 crashed on some Ubuntu Linux OS with a message containing
SSL: CERTIFICATE_VERIFY_FAILEDwhen using either the default --lookup ID or the --pubchem_names option. If the problem occurs then the environment variable BDG_GRADE2_SSL_DISABLE_VERIFICATION should be set (#540)
Fixed bug where the default
grade2 --lookupcrashes if the PubChem entry lacks a systematic name (#558).
When a CIF restraint dictionary with ambiguous chiral center(s) is used as an input, Grade2 now produces an output restraint dictionary where the chiral restraint volume is set to
bothfor the ambiguous centers rather than being arbitrarily assigned. (#560)
Fixed a problem where
DEBUGlogging output was wrongly produced when CIF restraint dictionaries were used as an input. (#561)
Extended bug fix #438 where long records cannot be read by Coot to SMILES strings. Now SMILES strings over 500 characters are not written to the CIF restraint dictionary. (#533)
v 1.3.0¶
- Distributed with
BUSTER 20221121
- Date
27 October 2022
New Features¶
A new option --lookup ID allows an external script to be invoked and look up details of a molecule from a corporate (or public) database and then run Grade2 to produce restraints for it. The environment variable
BDG_GRADE2_LIGAND_LOOKUPis used to set the location of the script. Please see https://gitlab.com/gphl/grade2_lookup_scripts for example scripts written in different languages and description of how to write you own lookup script.By default, if
BDG_GRADE2_LIGAND_LOOKUPis not set,grade2 --lookup CIDuses a script that downloads ligand details from PubChem https://pubchem.ncbi.nlm.nih.gov/ usingCIDthe PubChem compound identifier.Thanks to Christian Schleberger for suggesting this extension. (#519)
Grade2 will now write the systematic name of the ligand, if it is available, to the output CIF restraint dictionary. Systematic names for PDB ligands are automatically obtained from the input PDB chemical component definition. The --systematic option allows the systematic name to be manually set. For further details see the Systematic names section.
Thanks to Gilbert Bey for suggesting this extension. (#495 & #516)
The --pubchem_names option can be used to search online for the systematic name of a molecule by looking for it the PubChem database https://pubchem.ncbi.nlm.nih.gov/.
As the process involves uploading the SMILES string of the molecule to PubChem it should not be used for confidential ligands. To be extra careful, by default the option is deactivated please see --pubchem_names documentation for details of the activation process. (#529)
Added a new FAQ Security: does Grade2 upload any ligand information to public servers?
(#520).
The Grade Web Server has been updated and improved to run Grade2.
Please see the Grade Web Server chapter for more information. (&1)
The CCP4-extension CIF item
_chem_comp.groupis now set topeptidefor PDB chemical components that have _chem_comp.type set to eitherL-peptide linkingorD-peptide linking. In addition, for other inputs (such as SMILES, SDF or MOL2 file), if an alpha amino acid is recognized and atom IDs (N CA C O OXT CB) are set then_chem_comp.groupwill also be set topeptide. This enables Grade2 CIF restraint dictionaries to be used in Coot to replace protein residues with modified amino acids.Thanks to Chip Lesburg for suggesting this extension. (#471)
For PDB chemical components that have _chem_comp.type containing
saccharide,DNA LINKINGorRNA LINKINGthe CCP4-extension CIF item_chem_comp.groupis now set to eitherpyranose,furanose,DNAorRNA. For saccharides, the identification of eitherpyranoseorfuranoseis made using the full name for the ligand from _chem_comp.name. The improvement allows Grade2 CIF restraint dictionaries to be used for glycan and nucleic acid chains in Coot.Currently, no check for the chemistry of saccharides or nucleic acids is made for other inputs (such as SMILES). Please let us know if you would like this to be added. (#477 & #478)
A new option
--groupallows the CCP4-extension CIF item_chem_comp.groupto be manually set.Please see --group usage documentation for full details. (#479)
The new option
--aa_looseextends setting atom IDs to "exotic" amino acids. By default, only alpha amino acids with an unmodified amino group are recognized.--aa_looseextends recognition to N-modified amino acids, Aib-like amino acids with two beta carbon atoms, Gly-like amino acids, and beta amino acids. Please note, the option only works for input molecules that lack atom IDs (aka atom names) for instance a SMILES string or an SD file. For further details, please see the Atom Naming chapter.Thanks to Markus Rudolph, for suggesting this enhancement (&7).
The --PDB_ligand --rcsb option will now download information from https://files.rcsb.org/ligands/ in preference to Ligand Expo. This has the advantage that the https protocol is used and consequently is unlikely to cause firewall connection issues.
Thanks to Clemens Vonrhein for suggesting this improvement (#509).
Fixes¶
Fixed problem when Grade2 is supplied with a SMILES input that is then charged then atoms are often reordered during the charging process. This reordering can cause chiral inversions compared to the original input. The fix involves producing an initial restraint dictionary from the original SMILES string and then applying the charging routine to the initial restraint dictionary. This avoids reordering atoms and the the chiral inversion problems.
Thanks to Andrew Sharff and Matthias Zebisch for reporting the bug. (#470)
Remove misleading wedge indications of chirality from non-carbon atoms in SVG schematic 2D molecular diagrams. Now only carbon atoms will be marked as chiral in 2D schematics. For example, the PDB component VIA, once charged, previously had a schematic 2D diagram with wedges indicating that both a piperazine nitrogen atom and the sulfonyl sulfur atom are chiral:
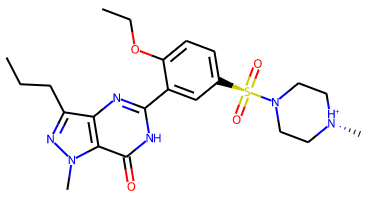
The misleading wedges have now been removed:
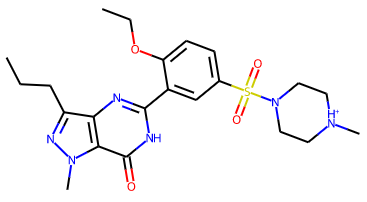
Thanks to Clemens Vonrhein for raising this issue. (#512)
Improve the WARNING message about amino acid atom labelling to include the option to turn off the feature --no_aa_labels. (#483)
Do not recognize fluoroglycine as a typical alpha amino acid with an amino group and a CB atom. (#488)
Support -ocif, with a single dash, as an alternative to --ocif option. This is more consistent with the original Grade program and should help to avoid confusion.
Thanks to Clemens Vonrhein for raising this issue (#500)
Fix bug where Grade2 terminates with an exception if supplied with an invalid SD file, instead providing clear error message. (#503)
Fix bug that
grade2_testsintermittently reports failure of testtest_problem_smiles_3d_coordinate_generation_raisesby using a doubly bridged naphthalene SMILES string that should be impossible to produce 3D coordinates for. (#506)
When dealing with PDB component containing element
X(such as https://www.rcsb.org/ligand/ASX) terminate tidily with a clear error message.Thanks to Clemens Vonrhein for raising this issue. (#510)
Properly handle PDB component containing deuterium as element
D(such as https://www.rcsb.org/ligand/TSD).Thanks to Clemens Vonrhein for raising this issue. (#510)
Fix problem with CCP4 energy type for hydronium ions (such as https://www.rcsb.org/ligand/D3O). (#511)
Do not terminate if the RDKit Minimize step has an error, instead give a WARNING and carry on to Mogul step. The problem encountered on PDB component I2I https://www.rcsb.org/ligand/I2I that can be handled after the fix.
Thanks to Clemens Vonrhein for raising this issue. (#513)
Terminate with a clean error message when asked to create a restraint dictionary for PDB component UNL https://www.rcsb.org/ligand/UNL that has no predetermined atoms.
Thanks to Clemens Vonrhein for raising this issue. (#514)
Grade2 can now read SD files produced by MOE whose terminating line
M ENDlacks theMcharacter.Thanks to Markus Rudolph for raising this issue. (#522)
v 1.2.0¶
- Distributed with
Grade2 Update Release 31 March 2022
- Date
31 March 2022
New Features¶
Grade2 will now by default, recognize a typical alpha amino acid with an amino group when supplied with an input that lacks atom IDs (aka atom names), for instance a SMILES string. If an alpha amino acid is recognized then the PDB-standard atom IDs (
N CA C O OXT CB) will be set for the main chain and beta carbon atoms and for the hydrogen atoms that they are bonded to. For further details, please see the Atom Naming chapter.If you prefer for the renaming not to happen, then the new Grade2 command-line --no_aa_labels option turns it off, leaving standard numerical order based atom IDs.
Note that, currently, no alterations are made if the input file specifies atom IDs (for example CIF restraint dictionaries and most MOL2 files).
Please let us know if you would like this feature extended, for instance to set PDB-style Greek letter remoteness IDs for side chain atoms beyond
CB.Thanks to Thierry Fischmann and Chip Lesburg for suggesting this extension. (#234)
A new option --ocif is introduced to set the full filename for the CIF restraint dictionary. This allows the specification of the exact filename to be used for output. It is most useful when used with the --just_cif option. Thanks to Steven Sheriff for suggesting this option. (#447)
Fixes¶
Grade2 should now deal with MOL2 files of charged molecules that have partial charges for atoms. To correctly identify the chemistry of a molecule the formal charge of each atom is required. This information is not stored in MOL2-format if partial charges are defined (the CSD-convention for MOL2 files is to use the partial charge field to store the formal charge). Grade2 now uses valency considerations to reconstruct the atomic formal charges if necessary. The fix has been tested with OpenBabel MOL2 files and copes with carboxylic acids, amines, imidazoles, nitro groups, azido groups, tetrazolates, isocyano groups, sulfanium groups, phosphonium and borates. Please let us know if you find a chemical group that causes problems. Thanks to Steven Sheriff for bringing the problem to our attention. (#444, #446 & #448)
Grade2 should now correctly handle MOL2 files that use bond type
arfor carboxylate groups. The CSD normalisation method can make a mistake when standardising the bonding of the group. Grade2 will now correct which oxygen atom carries the formal negative charge. Thanks to Dirk Reinert for reporting this bug. (#462)
Fixed problem whereby Grade2 restraint dictionaries could not be read by Coot because of long InChI records. The problem occurs because of long InChI records in the restraint dictionary, and it also occurs with CCP4-distributed restraint dictionaries. Currently, the CCP4 MMDB library (against which Coot is linked) places a line length limit of 500 characters, despite the IUCR CIF specification allowing lines of up to 2048 characters. We have let CCP4 know and the limit will be raised in a future CCP4/Coot release (by mmdb2 revision 56). From this release, Grade2 will no longer output long InChI records so there should be no problem in using Grade2 restraint dictionaries with older versions of Coot. Thanks to Steven Sheriff for bringing the problem to our attention. (#438)
If there is a problem with the RDKit chemical setup of a molecule read from MOL2-format Grade2 should now continue and produce a rudimentary fallback restraint dictionary rather than terminating with an error message. (#450)
If presented with an input molecule that has atom names (aka atom IDs) longer than 4 characters give a
WARNINGand do not output a PDB file. When producing custom atom names for molecules from SMILES avoid 5-character atom names where possible. (#454)
Fixed a problem where
DEBUGlogging output was wrongly produced when certain CIF restraint dictionaries were used as an input. (#453)
Fixed a problem where the message
WARNING: Proton(s) added/removedwas written toSTDERRwhen a ligand with charged atoms was processed. The message comes from the InChI generation routine and is nothing to be worried about. Now InChI generation warning messages are captured and available in the--debugoutput if they are of interest. Thanks to Dirk Reinert for reporting this bug. (#461)
Fixed a problem that the
grade2_utils --pdb_to_mol2script used bybuster-reportfailed when supplied with old CCP4 restraint dictionaries that contained chiral restraints with volumes such ascross2. Now the script logs aWARNINGabout invalid chiral volumes and continues. Thanks to Andrew Sharff for reporting this bug. (#463)
v 1.1.0¶
- Distributed with
BUSTER 20220203
- Date
01 February 2022
New Features¶
LIGis now used for the default residue name (aka PDB chemical component id or 3-letter code). Please see the FAQ on residue names for more information.Grade2 can now read an Grade CIF restraint dictionary as an
--ininput file. As Grade CIF restraint dictionaries lack atom formal charge (_chem_comp_atom.charge) records these are set zero when the restraint dictionary is read and care must be taken as this may cause the output molecule to be incorrect. The InChIKey is read from the Grade CIF restraint dictionary to enable a check that the stereochemistry matches. Please note that the bond orders from Grade restraint dictionaries can be incorrect. For further information, please see the FAQ: How can I use Grade2 to generate a restraint dictionary with atom names consistent with an existing Grade dictionary?. (#354 & #358)Grade2 can now read an eLBOW CIF restraint dictionary as an
--ininput file (as well as those from AceDRG, Grade and Grade2 itself). (#350 & #353)Known Issues and FAQs chapters added to this documentation (#313). The FAQs include "How can I run Grade2 if I only have a PDB file for the ligand?" and "How can I produce restraints for a ligand with a different protonation state or tautomer?" with a video demonstration. It is best to check the online versions of the chapters as these are frequently updated as new issues and questions come in:
Information about Mogul data libraries used is now included in the terminal output and the output CIF restraint dictionary in item
_gphl_chem_comp_info.mogul_data_libraries. The CCDC release periodic updates to the CSD through each year and these will be recorded. In addition, the use of Mogul information from in-house databases should be logged. (#368)A tool to produce MOL2 files for
buster-reportMogul analysis using Grade2 code has been produced. This is to avoid problems in chemical markup from coordinate file. This tool enables the chemistry of the ligand in Mogul analysis to be based on the CIF restraint dictionary used for refinement (after CSD standardization). Thegrade2_utilsscript option--pdb_to_mol2is used for the conversion. (#380 & #433)Grade2 now produces CIF restraint dictionaries with both electron-cloud and nucleus X-H bond restraints, avoiding requiring separate restraint dictionaries for the two use cases. The --ecloud option is retained to specify that the ideal coordinates for the ligand should use the electron cloud rather than nuclear distances. The BUSTER
refineoption-M Ecloudcan be be used to select the e-cloud model or-M HydrogenHybridModelthe hybrid model. (#431)Grade2 has been altered to produce a single plane restraint for each separate ring that is judged to be flat. Previously, planar rings were held flat by a number of four-atom planes. The --4_atom_planes option can be used to restore the previous behaviour. In practice, the change simplifies the restraint list but there is little difference in results. Please see the Treatment of Planar Groups chapter for more information. (#342)
The
_chem_comp_atom.type_energyfor hydrogen atoms is now set to proper context dependent values rather than being left asHfor all atoms. The information is required for BUSTER to setup non-bonded contacts properly distinguishing between polar, aromatic and other hydrogen atoms. (#406)The version information from the -V, --versions option has been extended to include information as to the location from which the CSD Python API is loaded. (#368)
The testing script
grade2_testshas been altered to output Grade2 version information. Thanks to Andrew Sharff for this suggestion. (#430)Added FAQ Grade2 says that the ligand matches an existing PDB chemical component. What should I do?. The FAQ is given in help messages by both the command-line and Grade Web Server interfaces. (#507)
Fixes¶
Fixed grade2_utils --pdb_to_mol2 bug in handling alternate conformations. (#422)
Improve error handling in the gelly optimization stage, so that on failure the full gelly output is reported and the restraint dictionary is then produced. (#412)
Fix bug where grade2_tests could fail with a message ending: PermissionError: [Errno 13] Permission denied /...some_path.../pytest.ini. (#404)
Clearer ERROR message is now produced if Grade2 is supplied with an invalid SMILES string. (#399)
Clearer ERROR message is now produced if Grade2 has a problem in 3D coordinate generation. The ERROR message now refers user to the FAQ https://gphl.gitlab.io/grade2_docs/faqs.html#xyz-generation-error . (#378)
Improved procedure for getting the CSD Python API directory from the CSDHOME when this has a symbolic link. (#336)
Work around developed that allows running Grade2 to run on MacOS with latest CSD Update Release 2021.2 (September 2021). There are on going issues with C-library duplication in CSD Python API that previously prevented Grade2 working. We have let the CCDC know about the C-library duplication. (#390)
Before updating your CSD installation, please check the online version of the Grade2-CSD compatibility page: https://gphl.gitlab.io/grade2_docs/csd_compatibility.html that is updated once new releases are evaluated.
Sorted bug in the --big_planes option. The --big_planes option merges smaller planes into as large a single plane as possible. This was done ignoring the sigmas (standard deviation of the out-of-plane distance) for individual restraints when merging. Grade2 places weak planar restraints on torsion angles that from CSD have a moderate preference for planarity and these were incorporated into a big plane. Weak planes (those with a sigma above 0.020 Angstrom) are now not incorporated into big planes. For further details, please see the Treatment of Planar Groups chapter. (#342)
Sorted bug where given an input MOL2 file containing atom names with lower case letters, Grade2 went on to produce a restraint dictionary with unaltered atom names. BUSTER expects atom names to be upper case. Now if atom names with lower case letters are found they are converted to upper case and warning messages are given, for instance:
WARNING: input has atom names with lower case letters: Br1 Cl1 WARNING: converting lower case atom names to upper case
Thanks to Dirk Reinert for reporting this bug. (#324)
Fixed bug that for the MacOS version update to CSD release 2021.1 (July 2021) caused Grade2 to crash with an
ImportErrormessage. (#331)
Fixed the final terminal output Suggestion: to view/edit the restraints, use one of the commands: to give correct commands if the --out option has been used to alter output filenames. (#333)
Sorted bug where grade2 will fail if the environment variable
PYTHONPATHis set. Thanks to Yong Wang for reporting the problem. (#349)
Grade2 can now read CIF restraint files where _chem_comp_atom.charge is supplied as a floating number rather than the standard integer number. Request from Andrew Sharff to support reading ligands restraint from PanDDA analysis of BAZ2B screened against Zenobia Fragment Library. (#353)
Fixed bug where
grade2 -checkdepsoption failed to give informative ERROR message when the CSD Python API installation was incomplete. Thanks to Vito Calderone for reporting the problem. (#394)
v 1.0.0¶
- Distributed with
BUSTER 20210716
- Date
14 July 2021
New Features¶
First public release of Grade2.
Internal pre-releases:¶
v 0.1.15¶
- Date
14 July 2021
New Features¶
Terminate with a clear error message if an attempt is made to run
grade2on a CentOS 6 system. (#301)First draft of the Documentation "Charging" chapter. (#274)
Fixes¶
Workaround to give exit status 0 if the Grade2 run is successful but where there is a
Segmentation faultorstd::bad_allocon shutdown. This should mean that exit status ofgrade2should reliably indicate success or failure. (#300)Improvements in the Documentation "Outputs" chapter. (#261)
Fixed minor bug where the first information line about
$CSDHOMEproduced by thegrade2script was not indented by a space. (#293)grade2_testsskip the test forEL9restraint dictionary generation with Mogul as it takes 55 seconds. (#294)Do not output final suggestion if
--just_cifoption is used (because the suggestions given require the PDB file). (#297)Fix many typos in documentation. (#303 & #305)
v 0.1.14¶
- Date
06 July 2021
New Features¶
The default output PDB chemical component id (aka residue name or 3-letter code) is now
L_1rather thanXXX. The use of an underscore ensures that there is no conflict with the id's of existing PDB components. (#273)The names of the files output by
grade2have been altered to make their contents clearer. In particular, the principal output restraint dictionary is now namedL_1.restraints.cif(as CIF-format is used for many types of data). The molecular diagram filenames start withL_1.diagram., whereas 3D coordinate filenames beginL_1.xyz.. (#266)The
grade2 -hhelp message has been improved. The Help & setup arguments are now listed as a separate group. All argument descriptions have been shortened with detail now given in the Documentation "Usage" chapter. (#256)All
grade2terminal output messages now start with a space, following a request from Clemens. This is for consistency with other BUSTER package programs and allows the distinction between program-produced and system messages. (#282).At the end of a
grade2run a suggestion for running Coot or EditREFMAC to view/edit the restraints is now made. (#268)Add
Normal termination (N sec)to the end of terminal output, giving elapsed seconds, following a request from Clemens. Also include the elapsed time information in restraint dictionary as CIF item_gphl_chem_comp_info.elapsed_seconds. (#281)If an input SDF file has 2D coordinates a
WARNING:message that XYZ coordinates are generated (#288).Improved error handling for PDB chemical components that lack complete ideal or model coordinates (a current example is
T0D). These cases will now terminate with the line:ERROR: the PDB CCD lacks complete ideal or model coordinates: cannot proceed.
Note that having incomplete coordinates often indicates that there is a problem with the chemical markup of the PDB component. (#61)
Fixes¶
Fixed bug in the
gellygeometry optimization chiral restraints setup that caused serious distortions for some chiral centers. (#265)The
-P PDB_ID, --PDB_ligand PDB_IDinput option will now convert a lower casepdb_idto uppercase, following Claus' suggestion. (#264)Fix bug where occasional atoms on phenyl rings next to bulky groups were not set planar (for example PDB ligand GVV https://www.rcsb.org/ligand/GVV atom
C9). (#270)Fixed bug where flat PDB ligands (such as QBK https://www.rcsb.org/ligand/QBK) failed with "cannot load RDKit coordinate as not 3D" exception. (#287)
Fixed bug where a Kekulization problem caused failure to produce a rudimentary fallback restraint dictionary for some PDB ligands containing metal atoms. The fix allows production of fallback restraint dictionaries for ligands such as X8P https://www.rcsb.org/ligand/X8P . Note that the rudimentary fallback restraint dictionary is based on the input coordinates from the PDB CCD where Mogul information is not available. (#286)
Fixed bug where the output log became scrambled when dealing with PDB CCD cif file input that had model rather than ideal coordinates (such as
TPP). (#290)
v 0.1.13¶
- Date
02 June 2021
New Features¶
Improvements in the Documentation "Usage" chapter. (#256)
Reduction in the size of the Grade2 distribution by removing unnecessary files. (#258)
v 0.1.12¶
- Date
28 May 2021
New Features¶
Once the restraints have been finalized, a final 'ideal' set of coordinates is produced by geometry optimizing the current coordinates with the restraints. The stand-alone gelly executable is used for the optimization to ensure compatibility with BUSTER refine. The optimized conformation is written to the restraint dictionary CIF file, PDB, SDF and MOL2 files. (#38 & #251)
The documentation section "Installation and Testing" now explains how to configure and test Grade2. (#250)
The configuration of Grade2 has been streamlined with clearer advice in error messages. A new optional environment variable
BDG_TOOL_CSD_PYTHON_APIhas been introduced for cases where there has been creative use of symbolic links in the location ofCSDHOME. (#250)
Fixes¶
Fixed bug that caused the grade2 0.1.12 rc1 Linux version to crash out with FileNotFoundError: ... screen_final.txt' message. (#253)
v 0.1.11¶
- Date
05 May 2021
New Features¶
Plane restraints are now used for torsions that are detected to have a strong trans or cis preference in Mogul analysis. Previously, 1-fold torsion angle restraints were used, like in old versions of Grade. 1-fold torsion restraints preclude flipping to the other rare conformer as well as not working well in recent versions of Coot. The plane restraints have an .id starting trans- or cis- so that the conformational preference information could be utilized downstream. (#240)
Restraints are now defined for torsions where there is a preference for a planar conformation but where steric interactions interfere. An example of this is provided by folic where a carbonyl group is attached to a phenyl ring and is most commonly found pushed out the plane. (#241)
For plane restraints the standard deviation for the out-of-plane distance (also know as the sigma) is now based on analysis of Mogul+ data for both ring and non-ring restraints. In practice, this means that ring planes will be held tighter than non-ring planes. (#241)
Fixes¶
The chemistry of nitro groups is now altered to the CSD convention before Mogul geometry analysis is performed. The MOL2 coordinate file output uses the CSD standardised bonding, including for nitro groups. (#229)
Sporadic bug in cython version that caused Mogul torsions results to be ignored traced to NamedTuple alteration, and hopefully fixed. (#243)
v 0.1.10¶
- Date
23 March 2021
New Features¶
grade2implement the command-line option-e, --ecloudto use electron-cloud distances for bonds to hydrogen atoms that are adequate for X-ray refinement. This option is based ongrade -ecloudand the same bond ideal distances and sigmas are used. (#37 & #220)
Fixes¶
Fix bug that chiral restraints involving a hydrogen atom could be produced (planning#5).
v 0.1.9¶
- Date
02 March 2021
New Features¶
The Grade2 command line option
--no_moguloption has been removed to be consistent with Grade. This means that CSD must be installed to use Grade2. (#204 and #212)Grade2 command line option
-checkdepsadded to be consistent with other BUSTER tools. The-checkdepsoption checks that CSD Mogul is accessible through the CSD Python API and works properly. Likegrade -checkdepsas part of check the ideal bond angle for carbon dioxide is found from a CSD mogul check.Started writing user documentation for Grade2. The documentation, in HTML and PDF formats, is included with BUSTER and can be found in the directory
$BDG_home/docs/grade2. The documentation includes this changelog. (#203, #216)The update process for the store of PDB chemical components InChiKeys has been automated to run every Wednesday after the weekly wwPDB release. This means that store should be up-to-date whenever Grade2 is released. (#210)
Fixes¶
Fix bug where Grade2 run through the distributed shell wrapper gave a exit status of 0 (success) when an error occurred. (#193)
v 0.1.8¶
- Date
02 February 2021
New Features¶
The miniconda environment that will be used to distribute Grade2 to users now has Grade2 installed in a binary form produced by cython. This means the Grade2 Python source code will not be distributed and so is protected from "prying eyes" and tinkering. (#169, #174 and #178)
The use of cython can be confirmed by using the
grade2option-V, --versions:$ grade2 --versions using CSD from $CSDHOME=/Applications/CCDC/CSD_2021/ grade2 0.1.8 (2021-02-02), RDKit 2020.09.1, Mogul 2020.3.0, CSD 542, csd_python_api 3.0.4 loaded from /Users/osmart/GPhL/BUSTER_snapshot_20190607/.mc/lib/python3.7/site-packages/grade2/*.cpython-37m-darwin.so PDB components InChiKey store last modified date: 2021-01-22
If a binary cython version is used then
loaded fromwill end in.soand containcpython
An automated procedure to produce the
conda_packtarballs that will be used to distribute Grade2 with the BUSTER installation has been developed. The procedure uses a GitLab CI/CD pipeline. The delivery process is run whenever a git tag is created, for instance, by making a GitLab release of the grade2 project. Separate installation tarballs are created for both Linux and macOS. (#177)
Once Grade2 is installed it can now be tested using the command
grade2_tests. This will run over-300 unit and functional tests using the pytest testing framework.grade2_testsprovides a quick way to ensure that a Grade2 installation works as it should, including that the CSD Python API loads and behaves as expected. (#183)
Fixes¶
Fix the WARNING message given if molecule is charged to give the correct
-N, --no_chargingoption and to be more readable. (#163)If a rudimentary fallback restraint dictionary is produced for from MOL2 coordinates the bond and angle restraints that are set from the input coordinates will have source
input_mol2_coords. (#149)Fix PDB output for cases where residue name (aka chemical component id) is not 3-letters so it is correctly column formatted. In addition restraint CIF dictionary item
_chem_comp.three_letter_codeis now truncated to the first 3 letters of_chem_comp.id. (#147)
v 0.1.7¶
- Date
12 January 2021
New Features¶
Procedure to distribute Grade2 with the BUSTER installation has been developed. Currently, this involves unpacking a tarball containing a miniconda environment produced by
conda_packand some helper scripts. For details contact Oliver. (#160 and #164).
Fixes¶
Fixed where CSD_PYTHON_API location setting failed if
CSDoccurred more than once in the CSDHOME path. (#167)
v 0.1.6¶
- Date
24 December 2020
New Features¶
Grade2 can now work using a run time import of the CSD Python API from the CCDC miniconda Python environment that is distributed with the CSD. Please note that this is likely to be the way that grade2 is included in the BUSTER distribution as it avoids update problems and redistribution of CCDC software. (#155)
Grade2 now checks the InChIKey of the input molecule against a store of those from the wwPDB chemical components definitions (wwPDB CCDs) https://www.wwpdb.org/data/ccd . This provides similar information to the
recognise-compoundfeature of Grade, with improvements such as detection of tautomers. For example, theCHECKuser output when generating restraints for PDB ligand 2D3:$ grade2 --PDB_ligand 2D3 (((output omitted))) CHECK: Check the molecule's InChiKey against known PDB components: CHECK: Exact match to PDB chemical component(s): CHECK: 2D3 https://www.rcsb.org/ligand/2D3 "methyl 3-isoxazol-5-yl-5-methyl-1H-pyrazole-4-carboxylate" CHECK: XQK https://www.rcsb.org/ligand/XQK "methyl 5-isoxazol-5-yl-3-methyl-1H-pyrazole-4-carboxylate" (((output omitted)))
shows that 2D3 ligand has a tautomer XQK in the wwPDB CCD. The output includes the RCSB URLs for each ligand as this is useful to help the user examine the hit(s). The check is made before time consuming step as this makes it more likely for the output to be read. The
CHECKoutput is included in the output restraint CIF in the items_gphl_check_inchikey_pdb_ccd.text. The wwPDB CCDs store used is provided in the separate repo that will be updated on a weekly basis. (#104)Improvement in the logging information that Grade2 provides about a PDB component to include the RCSB and PDBeChem URLs for the molecule. In addition, the all upper case molecule names used for old components are now reformatted for readability. Using component
468as an example:$ grade2 --PDB_ligand 468 (((output omitted))) Collected PDB chemical components definition for PDB id 468 from: http://ftp.ebi.ac.uk/pub/databases/msd/pdbechem_v2/4/468/468.cif Molecule name: "(3S)-N-(3-chloro-2-methylphenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide" For more information about "468" see: ---- https://www.rcsb.org/ligand/468 ---- https://www.ebi.ac.uk/pdbe-srv/pdbechem/chemicalCompound/show/468 (((output omitted)))
The URLs provided
should help in quickly looking up information about the component. (#151)
Fixes¶
The csd_python_api version number is included along side Mogul and CSD version numbers both in user output and in the output restraint dictionary CIF file as item
_gphl_chem_comp_info.csd_python_api. (#154)Switch to using the latest PDBeCIF parser directly available from pip. This simplifies the installation process removing the need to separately install PDBeCIF. (#157)
The installation section of README.md has been updated (#158).
v 0.1.5¶
- Date
27 November 2020
New Features¶
Grade2 has a new option
-s, --shelxto produce SHELX restraint.dfixfiles. If specified two additional files will be created with the suffices.dfixand.with_hydrogen.dfix. The former file has restraints excluding those to hydrogen atoms. The actual filenames will be depend on theOUT_ROOTthat can be set with the-o OUT_ROOT, --out OUT_ROOToption if the default is not suitable. (#26)
Grade2 will now try to produce a rudimentary fallback restraint dictionary for PDB ligands where there is an RDKit sanitization problem. This can occur in cases where the PDB Chemical Components Definition has valency problems or for problematic groups such as carborane. The rudimentary fallback restraint dictionary will be based on input coordinates where Mogul information is not available. (#134)
Improve treatment of metal-containing PDB ligands to recognize dative bonds and run Mogul against CSD organometallics. Restraint dictionaries for compounds such as heme (HEM) are improved. (#135)
Note that there is still a limitation that the UFF force field setup does not work for transition metals because of RDKit limitation. Much future work is required to treat metals properly.
Fixes¶
The ligand name is now included in the user log output for PDB ligands. (#137)
Grade2 is now hard coded to only create chiral restraints with a central carbon atom, so nitrogen atoms that RDKit recognises as chiral will no longer be affected. (#120)
v 0.1.4¶
- Date
05 November 2020
New Features¶
Grade2 now provides improved logging of the InChI comparison. When an InChI is available from the input (for instance for PDB ligands using -P PDB_ID) this is compared to the InChI for the RDKit molecule generated. If there is a match then this is noted in the output log as this is a good indication that the stereochemistry of molecule has been correctly setup. If there is a mismatch then a WARNING message is produced. Information about the InChI comparison is also provided in the output CIF restraint file in items _gphl_chem_comp_info.input_inchi* to allow machine reading. (#124)
A script
pdb_ideal_mol2_generatorhas been added that produces a MOL2 file for a given PDB ligand from the PDB Chemical Component Definition using Grade2 input parsing to RDKit and the CSD Python API. For help on using the script use the-hoption. Please note this is only likely to be useful to developers for test and may be removed before release to users. The grade2 option-P PDB_ID, --PDB_ligand PDB_IDshould be used for to generate restraint dictionaries for PDB ligands. (#126)
v 0.1.3¶
- Date
02 November 2020
New Features¶
Grade2 now supports molecule input from a CIF-format restraint dictionary produced by Acedrg or Grade2 itself. Unfortunately because of incomplete information it would be difficult to support reading of Grade restraint dictionaries. (#105)
Grade2 now outputs SDF and MOL2 format files for the molecule in addition to the PDB format file. The SDF and MOL2 files have the advantage of explicitly including bonding and atom formal charge information. The MOL2 file is written by CCDC routines and represents the chemistry supplied for Mogul analysis. (#116)
The output CIF-format restraints dictionary produced by Grade2 has been extended to include information about bond aromaticity. The CIF item _chem_comp_bond.aromatic is used following the practice of Acedrg. The information presented is from the RDKit_aromaticity_model. It should be noted that there are a number of different models of aromaticity, that can lead to different results for fused and multi-ring systems as demonstrated in the OpenEye OEChem Toolkit page on aromaticity_perception. For this reason, procedures based on aromaticity perception should be undertaken with caution. For this reason, Grade2 does not use aromatic information internally. (#122)
Fixes¶
SMILES and InChi descriptors are now reported as CIF item _pdbx_chem_comp_descriptor in the output CIF-format restraint dictionary to conform this the PDB Exchange Data Dictionary. (#115)
Bug where SMILES files containing just the SMILES string and no names caused a crash has been fixed. (#118)
Fix bug where charging adding hydrogen atom starting from MOL2 input caused crash "ZeroDivisionError: float division by zero". (#119)
Fix bug reading acedrg CIF restraint dictionary from MOL2 start that lacks a _pdbx_chem_comp_descriptor information for SMILES and InChIKey. (#121)
v 0.1.2¶
- Date
19 October 2020
New Features¶
Grade2 can now handle file input from
smi(SMILES) file type. (#25)Grade2 can now handle file input from MOL2 (SYBYL) file type. (#24) Routines from the CSD Python API are used to input MOL2 files as the RDKit MOL2 parser has limitations. (#113)
Fixes¶
Grade2 can now handle monoatomic PDB ligands, like NA sodium ion. (#65)
Improve handling of problematic SMILES. If there is a problem in the initial coordinate generation will now retry using random coordinates. (#86)
Where PDB Chemical Component Definition has _chem_comp_atom.charge as '?" grade2 will now set the charge to 0 and issue a WARNING message (problem arose for PDB ligand QQ7). (#67)
Alter
grade2_utilscommand-line options to be consistent withgrade2. The option--compare IN_FILE2now checks that the fileIN_FILE2exists before opening. (#107 and #52)Error messages about CSD Python API and Mogul problems have been cleaned up and include suggestion of rerunning with -n, --no_mogul. (#60).
Grade2 now produces a sensible error message if supplied with a file that cannot be processed (#108).
Bug where non-zero _chem_comp_atom.charge was not set working starting from SMILES input has been fixed. (#109)
Charging carboxylic acid to carboxylate no longer assumes hydrogen atom specified second in bond. Fixes bug for CSD MOL2 INDPRA01 (#112).
v 0.1.1¶
- Date
05 October 2020
New Features¶
Grade2 can now handle file input from
molandsdffile types. (#23)Command line option
--itypeimplemented to allow user setting of the file type. By default, this is detected from the filename extension and file contents. (#23)Command line option
--nameimplemented to set_chem_comp.namename of compound. This will be displayed in buster-report. (#28)Command line option
--database_idimplemented to set a database_id. buster-report will provide a hyperlink for known PDB ligands. (#28)Command line option
-b, --big_planesimplemented to produce fused planes rather than lots of 4-atom planes. (#27)
Fixes¶
Grade2 will now produce logging output to STDOUT rather than STDERR. This is similar to original Grade and makes redirection of output much easier (#103).
v 0.1.0¶
- Date
23 September 2020
New Features¶
The project is now called "Grade2" rather than "Gorr". Command-line scripts for users are now called
grade2andgrade2_utils(#94). Command line options forgrade2have been revised in line with the "Grade2 Release Candidate Proposal Document". (#93 and #96)Charging common neutral groups such as carboxylic acids, phosphates and alkyl amines. By default if you supply Grade2 with a molecule that has a neutral carboxylic acid and/phosphate group this will be deprotonated to form charged carboxylate or phosphate ion. Conversely if you the molecule has an 'alkyl amine' (that is a neutral nitrogen atom bound to hydrogen atoms and/or carbon atoms that are connected to 4 other atoms) a proton will be added to it. This charges primary amino, piperidine, and piperazine groups. To turn off the feature then use the command line option
--no_chargingor-N. Please Oliver know if you would like for the list of groups to be charged to be extended. (#53)If Grade2 is supplied with a SMILES string that has ambiguous stereochemistry then the user will be warned and the resulting restraints will have the chiral restraint volume set to
both. In other cases of ambiguous stereochemistry command-line option--chirality_bothor-ccan be used to set chiral restraint volumes toboth. (#71)grade2_utilscan read restraint CIF files from CCP4 ACEDRG to facilitate comparison of restraints between Grade2 and ACEDRG. (#81)
Fixes¶
Planar atoms without full Mogul information now set from MMFF94s out-of-plane restraint rather than sum of bond angles (#84)
Chiral restraints no longer placed on phosphorous atoms. These restraints can cause distorted phosphate groups if the oxygen atom's atom_ids are not standard. Grade2 is now hard coded to only create chiral restraints with a central carbon atom, so phosphate groups will no longer be affected. (#75 and #120)
Fix bug where piperidine and piperazine ring nitrogen atoms wrongly set planar from Mogul results (PDB ligands 9JY and VIA) (#83)
Ideal bond angles not available from Mogul now taken from force field optimized values rather than the force field equilibrium value. This is get around cases where MMFF94 has bond angle restraints inconsistent with planar restraints like atom
N6of ATP. (#91)
Other Changes¶
This ChangeLog added (#80).
v 0.0.2¶
- Date
September 02, 2019
Fixes¶
Restraint CIF produced for PDB ligand CLF (FE8-S7 cluster) (#62).
gorr -PDB_ligand now retries the download 3 times after a wait of 0, 10, 40 seconds wait (#58).
Deal with PDB ligands lacking or incomplete ideal coordinates, for instance TDP (#55). Model coordinates will be used.