Direct use of weighted Quantum Chemical Energy for ligands in BUSTER refinement:
Introduction to to the method
- The idea adapts a protein simulation method called MM/QM where a quantum chemical method (QM) is used to represent a small ligand model within a protein that is modelled by a molecular mechanics type force field (MM). For more details see
- We are not the first people to adapt MM/QM for protein refinement. Prof K M Merz and co-workers have done a great deal of excellent work in this area see:
- The adaptation for protein refinement is to simply use a weighted quantum chemical energy for the ligand as part of the geometrical restraint function during refinement.
- expressing this as a picture:
- In terms of BUSTER refinement it simply means that an additional term is added to the objective function
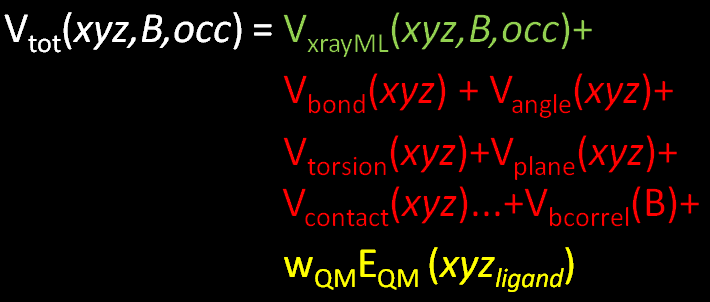
- as normal refinement minimizes the total function by adjusting the model parameters Cartesian coordinates of all atoms xyz, temperature factors B and atomic occupancies O.
- Eqm is the potential energy of the ligand as calculated by the quantum chemistry calculation and Wqm is a weight that can be adjusted (see tutorial).
- Note that the weighted QM contribution only depends on the coordinates of ligand itself.
- In terms of the refinement program itself BUSTER takes the approach of using an external separate helper QM helper program
- BUSTER writes a file with the coordinates of ligand, charge and quantum method to be used.
- BUSTER then invokes the helper application via a system call
- The helper executable then performs the quantum chemical calculation and provides the potential energy and its derivatives back and writes the results to a disk file.
- BUSTER reads the potential energy and its derivatives from the disk file. These are weighted and contribute to the overall objective function.
- although this is somewhat crude it is a common approach taken in many MM/QM applications.
- A helper application that uses fdynamo is included as part of the BUSTER distribution. This helper enables the use of the rapid yet reasonably accurate RM1 semi-empirical quantum method, see AutobusterLigandQMhelpers for more details.
- It is also possible to use more sophisticated ab initio or DFT quantum chemical methods, see AutobusterLigandQMhelpers.
- The approach taken here basically equates to using the quantum chemical procedure as a sophisticated restraint function for the ligand.
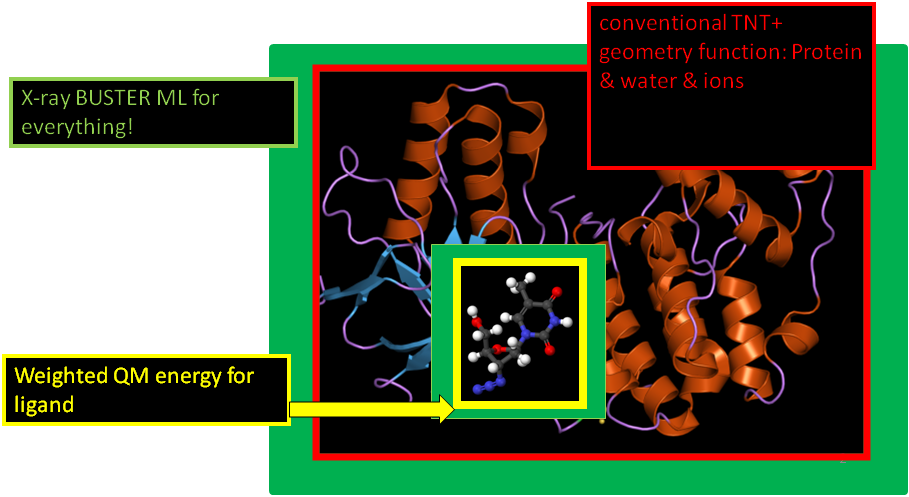
- The quantum chemical calculation is undertaken in vacuo with no contribution from the surrounding protein to the quantum calculation. In MM/QM terms this corresponds to the simplest 2-layer ONIOM approach. ONIOM is a technique developed by Gaussian for a good introduction see Gaussian Introduction to ONIOM web page
- The ligand atoms make a normal contribution to the BUSTER Maximum Likelihood X-ray calculation.
- Ligand atoms have normal non-bonded ideal contact to the protein and BCORREL coupling restraints on B factors. But other than this the ligand atoms do not have any normal bond, angle, plane, torsion or contact restraint to each other. In this implementation this is achieved by turning off or zero weighting all such interactions.
- To cope with the common situation where NCS means that there is more than one copy of the ligand bound to the protein the approach allows for multiple independent QM sets. This means that potential energy each copy of ligand is found by a separate invocation of helper executable.
Why use weighted-QM-energy as a restraint function for ligands?
- the weighted-QM-energy restraint function really provides a second opinion compared to conventional geometrical restraints. It is particularly useful for the situation where it is suspected that a particular conformation observed in a complex structure is due to the restraints used.
- Suppose the ligand stereochemistry is well represented by both a conventional restraint dictionary and the weighted-QM-energy restraint function. In this case complex structures obtained with two methods can be expected to have similar final ligand conformations.
- In addition the weighted-QM-energy restraint function can be used to assess strain energy and which conformation degrees of freedom are strained.
Using the weighted-QM-energy as a restraint function for ligands
Page by Oliver Smart 03 Feb 2011. Any questions regarding our software or this wiki should be directed to buster-develop@globalphasing.com