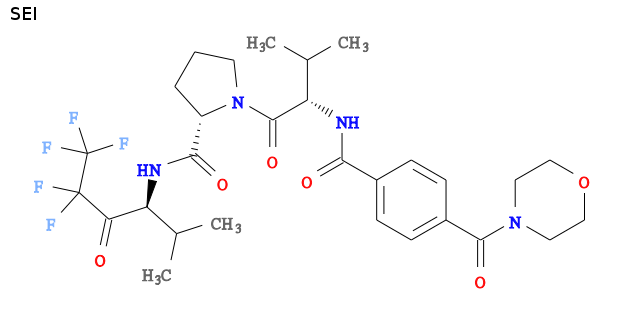
How to use grade to get BUSTER restraints for a covalently bound ligand, advanced example 1b0e/SEI
- The method is described in detail in GradeCovalentTutorial
- this page needs to be updated to work with ccp4 6.3.0 Please use the GradeCovalentTutorial method
- here we will apply to 1b0e, rcsb page on 1b0e
- the ligand is SEI, ligand expo on SEI
- Grade Web Server results on SEI
- my interpretation is that SEI binds covalently to the active site serine by forming a hemiketal link, see http://en.wikipedia.org/wiki/Hemiketal
- So following the method we need to use the ccp4i sketcher to form an extended ligand:
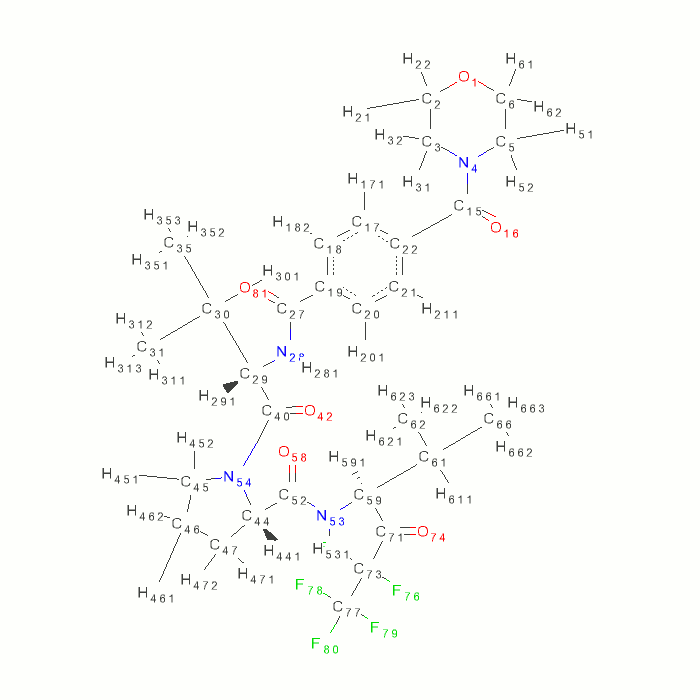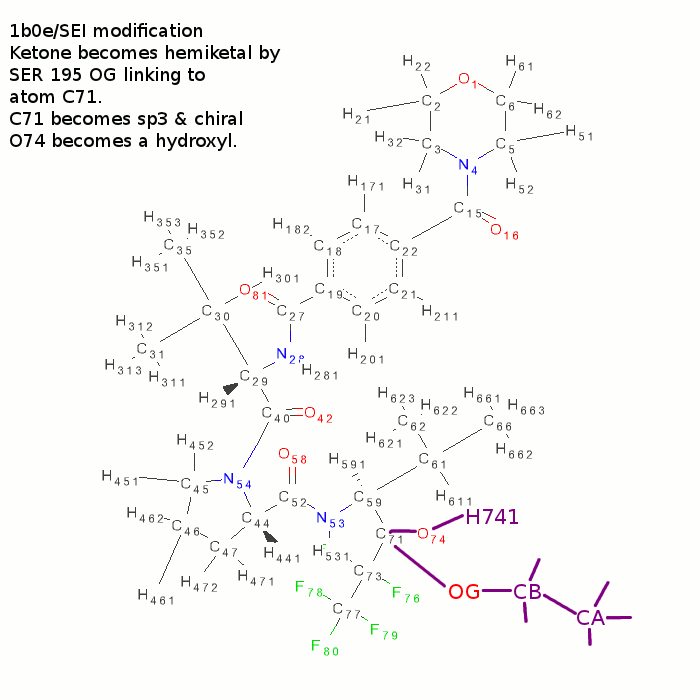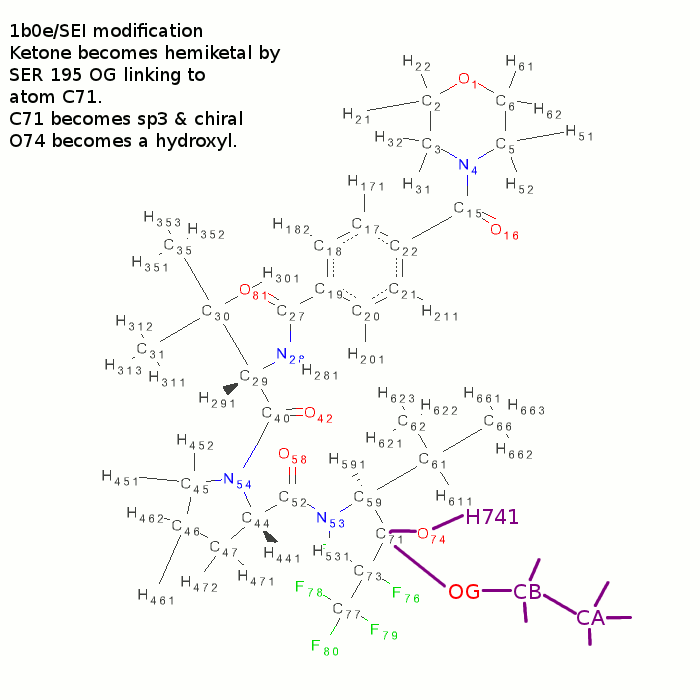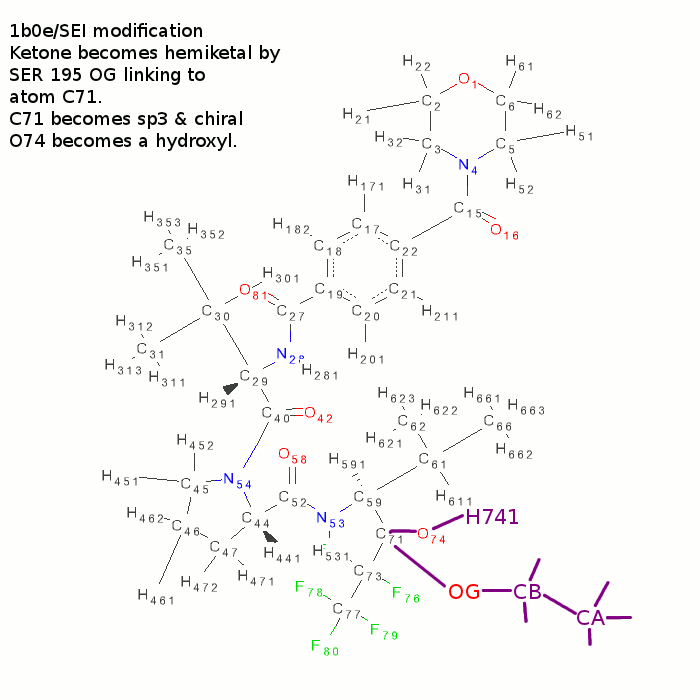
- use ccp4i sketcher.
- load SEI.grade_PDB_ligand.pdb
- change C71-O74 bond to a single
- Add H741 atom to O74.
- Add a chain of three extra atoms to C71
- Change first extra atom to an oxygen. Rename to OG, CB and CA.
- N.B. atom C71 becomes chiral - there is a 5 0/50 chance that the new chiral centre will be correct!
- Use the "File", "Create Library Description" and select the options "displayed structure" in the "Create description from" option. Use the unique identifier SEISER. Also select the "Regularise the structure with Refmac" and "load and display hydrogen atoms". The "Output coordinate file for idealised coordinates" is SEISER_libcheck.pdb
- convert the pdb file to mol2:
obabel SEISER_libcheck.pdb -O SEISER_libcheck.mol2
1 molecule converted
- Use the mol2 file as an input to Grade Web Server. Use residue name SEI, results of this:
- Then use perl script GradeCovalentTutorial/covalent_grade_create_linkdic.pl (file not found) to produce TNT link dictionary from the extended-ligand cif dictionary
./covalent_grade_create_linkdic.pl -icif grade-SEI.cif -odic grade-SEISER-link.dic -3let SER -aatnam OG -latnam C71
# covalent_grade_create_linkdic.pl
# use -man option to see full copyright information
# Converting dictionary grade-SEI.cif with refmacdict2tnt resulted in 329 lines starting GEOMETRY, for residue type SEI
# three letter code for amino acid that is modified is SER
# success identified 9 link type restraints and have written them to file grade-SEISER-link.dic
- The result is a link dictionary:
cat grade-SEISER-link.dic
# tnt dictionary to link amino acid SER atom OG
# to ligand SEI atom C71 using restraint type ZSEI
# produced by covalent_grade_create_linkdic.pl jiffy
CATEGORY_AT SER SER
CATEGORY_AT SEI SEI
CONNECT_AT SER OG SEI C71 ZSEI
GEOMETRY ZSEI BOND 1.382 0.030 OG +C71
GEOMETRY ZSEI ANGLE 105.9 3.0 OG +C71 +C59
GEOMETRY ZSEI ANGLE 115.0 3.0 OG +C71 +O74
GEOMETRY ZSEI ANGLE 103.8 3.0 OG +C71 +C73
GEOMETRY ZSEI ANGLE 121.6 3.0 CB OG +C71
GEOMETRY ZSEI TORSION 10000.0 1000000.0 +C59 +C71 OG CB
GEOMETRY ZSEI TORSION 10000.0 1000000.0 +C71 OG CB CA
GEOMETRY ZSEI CHIRAL 1 1 +C71 OG +C73 +O74
- It can be noted from the sigma's that the bond lengths and angles are based on RM1, this is because there is only one hemiketal containing CSD entry (FOVSUE).
- The pdb file 1b0e.pdb contains two links records for SEI and SER
LINK OG SER A 195 C71 SEI A 260 1555 1555 1.49
LINK OG SER A 195 O74 SEI A 260 1555 1555 2.04
- The first is correct but the second is in error and must be removed.
- Doing this and running hydrogenate:
hydrogenate -p 1b0e_correct_link.pdb -o 1b0e_cl_addh.pdb -l grade-SEI.cif
hydrogenate uses the 'reduce' tool; if you publish work which uses it,
please cite
Word, et. al. (1999) J. Mol. Biol. 285, 1735-1747.
For more information see http://kinemage.biochem.duke.edu
Producing CONECT records suitable for molprobity.reduce on SEI
Residues CA not hydrogenated; give dictionaries (with the -l dic1 dic2 ... option) to do this
- result 1b0e_cl_addh.pdb
- inspecting this reduce fails to place H741 the hemiketal hydrogen atom. This is probably because the hydroxyl hydrogen has to squeeze into the oxyanion hole and so there is no great position. The atom needs to be placed so did this manually in coot.
- result 1b0e_cl_addh_h741.pdb (file not found)
- Then run MakeLINK to create a sequence file.
MakeLINK -p 1b0e_cl_addh.pdb -o 1b0e_cl_addh.seq -d grade-SEISER-link.dic
- before running a full refine check that the restraints make sense for the covalently linked SEI - particularly the chiral centre C71.
refine -p 1b0e_cl_addh.pdb -m 1b0e.mtz -Seq 1b0e_cl_addh.seq \
-l grade-SEI.cif -l grade-SEISER-link.dic -d 1b0e_01_gradecovalent_maponly \
-M MapOnly > 1b0e_01_gradecovalent_maponly.log
- This results in a WARNING in the output log
WARNING: (chiral) Have 1 chiral atoms that are inverted:
WARNING: (chiral) A|260:C71(SEI)
- so by chance the link dictionary has set the wrong chirality for atom C71. To get around this it is necessary to produce a new link dictionary with the opposite chirality:
- change CHIRAL line by swapping the last two atoms so
GEOMETRY ZSEI CHIRAL 1 1 +C71 OG +C73 +O74
GEOMETRY ZSEI CHIRAL 1 1 +C71 OG +O74 +C73
- So finally we can refine with the good restraints for the link:
refine -p 1b0e_cl_addh_h741.pdb -m 1b0e_cv_corrected.mtz \
-Seq 1b0e_cl_addh.seq -l grade-SEI.cif -l grade-SEISER-link-correct-chiral.dic \
-d 1b0e_02_refine > 1b0e_02_refine.log
- results.
- 1b0e_02_refine.pdb
- 1b0e_02_refine.mtz
- The SEI ligand has the hemiketal link as desired:
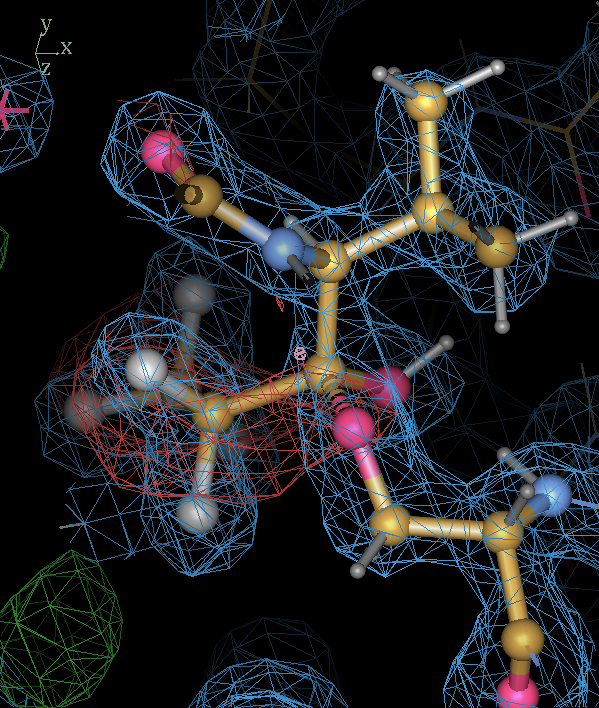
- The refined structure shows that there is a lot of difference density around the fluoroethyl group next to link. In addition the 2Fo-Fc splits in this region. The hemiketal hydroxyl hydrogen is not happy in the oxyanion hole. The hemiketal interpretation may not be correct! But in any case we have produced a model with reasonable restraints to test it.
Page by Oliver Smart original version 06 June 2012. Address problems, corrections and clarifications to buster-develop@globalphasing.com