This example, again using beta-secretase structures, illustrates a slightly more complex situation which requires specification of extra commands and changing defaults for pipedream.
For this tutorial, the reference structure is PDB id: 3NSH and the experimental data is taken from PDB id 3I25.
| 3i25.mtz | the structure factors as supplied by the pdb for 3i25, converted to mtz format |
| 3nsh.mtz | the structure factors as supplied by the pdb for 3nsh, converted to mtz format |
| 3nsh.pdb | the model coordinates as supplied by the pdb for 3nsh |
(A) Restraint dictionary generation
3i25 was soaked with a large and very flexible inhibitor, ligand code MV7.
grade2 -in MV7.smiles -r LIG
The structure that is being used as the "reference" in this case, 3NSH, is not a native structure but has a bound ligand (ligand code 957) in the active site.
grep -v " 957 " 3nsh.pdb > input.pdb
We are now ready to run Pipedream:
pipedream -hklin 3i25.mtz -xyzin input.pdb -hklref 3nsh.mtz \ -rhofit LIG.restraints.cif -rhothorough -d pipe1
The main output file summary.out indicates that the job has run as expected.
The initial refinement has been successful and Rhofit has "successfully" fit the ligand into each of the 3 protein chains. The "best" solution has been post-refined with very good statistics - R = 16.9%, Rfree = 20.0%.
Again, the real test is whether or not the ligand has been correctly fit and refined.
The image below shows the ligand in chains A, B and C.
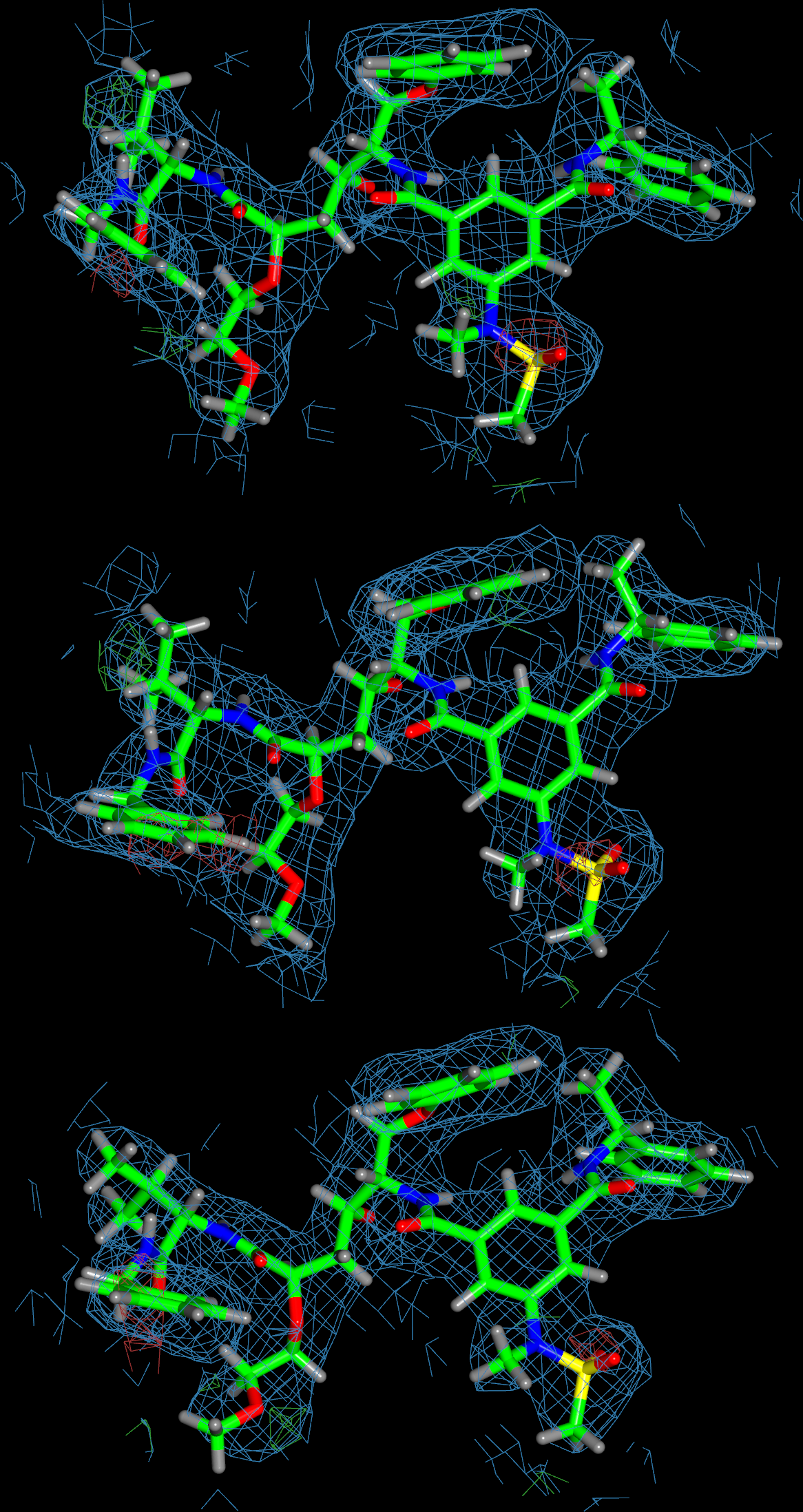
The orientation of all 3 copies is similar and in each case the ligand fits the density (with little difference density present).
Furthermore, they are a good match to the original 3i25 structure.
Look at the buster-report output:
firefox report/index.html
The ligand analysis is good with no outliers and the molprobity analysis also looks good. Again, there is clearly some room for improvement in the structure - this will need manually correcting in Coot and a subsequent re-refinement.