Pipedream Tutorial 3
Introduction
A common practice to reduce the number of x-ray experiments in the first phase of pursuing a fragment based screening strategy is to soak crystals with multiple compounds, rather than screen fragments individually.
This typically involves soaking crystals in cocktails ranging from 2 to around 8, small (hence usually weakly binding) and importantly, shape diverse fragments and then analysing the difference electron density maps to determine which if any of the components have bound to the target.
Pipedream currently provides basic support for this scenario.
Before proceeding with the tutorial, we should highlight a number of points which need to be carefully considered when using Pipedream to look at structures containing small, weakly bound fragments (whether soaked as cocktails or individually). Given that the compounds are small (and generally weakly bound), we are looking for similarly "small" difference density features and need to be mindful of the increased risk of mistaking difference density features that may be the result of conformational change, disorder, incompleteness of the model (or indeed data) or simply "noise" from genuine compound peaks - so called false positives.
- The choice of "reference" structure to use as the input to Pipedream is very important. That is true whatever the size and binding affinity of the ligand, however the smaller and weaker the fragment, the more critical the choice of starting model becomes. The reference structure should be as near identical to the fragment bound structure as possible, with the obvious exception of the fragment. If ligand binding is known to result in conformational changes, even if these are limited to shifts in amino acid sidechains in and around the ligand binding site, then you would be well advised to use a ligand bound structure (of course with the ligand(s) removed from the model) as the reference structure, in preference to an apo model. If the target also contains prosthetic groups and/or buffer components that are observed binding in sites other than the expected fragment binding site, then these SHOULD be included in the reference structure.
- By default, Pipedream will currently remove all waters from the reference structure and re-build the solvent model in the course of refinement. However, if the solvent structure is observed to be stable and largely conserved, then it makes more sense to retain the existing water model. This will reduce the sigma levels in the initial maps and give the refinement process an early head start.
In order to keep existing waters from the reference structure the following points MUST be followed:
- Water molecules (and indeed ANY hetatms) to be retained MUST be assigned the same chain identifier as the protein chain to which they are associated. Any waters with a different chain identifier will be discarded!
- Pipedream must be run with the -keepwater keyword.
- Additionally, where Pipedream is run with small fragments, it may well be advantageous to also use the -nowateradd keyword in combination with retaining existing water structure, to tell Pipedream that it should not attempt to update the existing water model in refinement (either by adding or removing waters). The potential downsides of the use of this option are that:
- The ligand binding site is not excluded from the bulk solvent correction in BUSTER. If your fragment is poorly bound or has low occupancy, its electron density may be "washed out" / degraded by the bulk solvent correction to the point where ligand binding is missed - false negatives.
- If used in conjunction with -keepwater, and the water structure is not well conserved between the reference structure and the experimental structure, you are including atoms that are not present in the model (or are in the wrong place) and preventing BUSTER from effectively correcting them. You are thus increasing the sigma levels in the map and make identification of fragment binding more difficult.
This tutorial uses some trypsin structures to illustrate how to use Pipedream with cocktails of small fragments. A series of trypsin structures soaked with a series of compounds, both individually and in cocktails, is described by:
Yamane, J et al. (2011), J. Appl. Cryst, 44, 798-804.
All of the resultant structures are available in the PDB.
For this tutorial we will use the following structures/data:
3rxt was soaked with a cocktail of two compounds (ligand codes SZ4 and 2AP).
(A) Restraint dictionary generation
- generate grade2 restraint dictionaries for ligands S24 and 2AP from their smiles descriptions as follows:
grade2 'COc1cccc(CN)c1' -r L01
grade2 'Nc1cccc[nH+]1' -r L02
(B) Pipedream run
Before we can run Pipedream, we need to "prepare" the reference structure.
3rxe is not an apo structure, but has benzamidine bound in the binding site. So, before using 3rxe we need to remove the benzamidine from the model. Also, as this is a high resolution structure with a stable water structure, we will keep the water model rather than have Pipedream discard and rebuild it. But we want to ensure that there is no solvent encoaching on the ligand binding site.
- View 3rxe in Coot and centre on the benzamidine (A5). Delete the benzamidine and waters in the immediate vicinity of it (around 6 in this case). In this structure, there is a single chain in the asymmetric unit. Confirm that the built waters have the same chain identifier as the protein. Save the updated coordinates for use as the input file for Pipedream - input.pdb.
We are now ready to run Pipedream.
Run Pipedream with the following command:
pipedream -hklin 3rxt.mtz -xyzin input.pdb -hklref 3rxe.mtz \
-keepwater -nowateradd -rhofit L01.restraints.cif,L02.restraints.cif \
-d pipe &
- The restraint dictionaries for each component of the cocktail are specified as a comma separated list (with no intervening spaces) as the argument for the -rhofit option.
- Use of the -keepwater -nowateradd options tells pipedream that the input water structure is to be kept as is.
Note
You should also consider the following points when running Pipedream with cocktails:
- Currently, it is not possible to identify specific binding sites for Rhofit. Run with default options, Rhofit will pick what it considers to be the best difference density feature in which to try to fit the ligand(s). As mentioned above, with small, weakly binding fragments, there is an increased risk of Rhofit choosing a feature away from the expected binding site, resulting in a false positive. Of course it could be indicating genuine alternative binding sites. The solution to this is to use the -allclusters option in Pipedream to tell Rhofit to fit the ligand to all possible clusters. Of course this will increase the time the job will take to run, however it has the advantage that all potential binding events will be analysed. The results will need to be closely analysed to decide which if any of the solutions are genuine. This is a determination that only you can make.
- As mentioned, only the "best" solution from Rhofit is post-refined. If Pipedream is run without -allclusters and has selected the wrong cluster, then more cpu time may have been needlessly used on post-refining an incorrect or unwanted solution. In addition to -allclusters, also specifying -postref will further increase the cpu time required, but has the definite advantage that the electron density maps, both 2fofc and fofc, after refinement should give much greater discrimination between a potential binding solution and a false positive. Use of -postquick instead of -postref (to perform a faster, more curtailed refinement) would be a reasonable option to save time in this instance.
(C) Analysis
Now, we will look at the output and the results.
- The primary "standard" output from Pipedream is in summary.out.
- You will notice that the Ligand Fitting and Post-refinement section is now repeated 2 times - once for each of the two components of the cocktail.
- It is IMPORTANT to note here, that just as Pipedream cannot determine which of the potential solutions that Rhofit has built for an individual compound is correct, where Pipedream is run with a cocktail, it similarly cannot and will not try to determine which, if indeed any, component(s) of the soaked cocktail are bound. It simply presents the results of trying to fit each component in turn. You must determine what the correct outcome of the experiment is yourself.
- In particular, the scores from Rhofit may not be directly comparable, especially if the components differ significantly in size/number of atoms. In the case of comparing different ligands, the most appropriate score to look at though is the density correlation coefficient.
Look at the Rhofit and post-refinement results in the summary.out file.
- Look at the R / Rfree after post-refinement. They are pretty similar to each other. R/Rfree are not particularly sensitive to small aand highly localised differences in the model and do not constitute good metrics to evaluate which, if any, of the ligands is correct.
- In this case, the density correlation coefficients calculated by Rhofit for the 2 solutions are more interesting. In this case, ligand L01 has the higher CC.
- Look at the output of buster-report for each of the 2 solutions and look at the "ligand report tab"
firefox report-grade-(L01,L02)/index.html
- In the middle column is a 2fofc density correlation coefficient (calculated from BUSTER). In this case, the pattern also agrees with the CC calculated by Rhofit - L01 > L02
These would suggest that L01 is correct.
Now, look at the Rhofit solutions graphically:
visualise-rhofit-coot rhofit-grade-(L01,L02)
and look at the difference density that Rhofit was trying to fit into and the solutions it found.
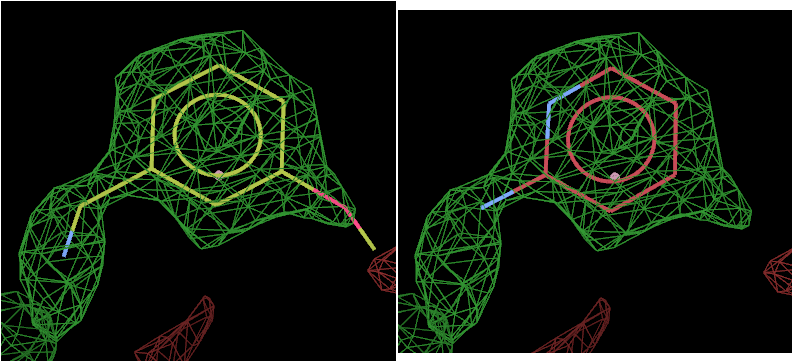
The above picture shows the top solution from L01 on the left and L02 on the right.
The density strongly suggests that L01 has bound.
Now look at the output maps from post-refinement:
visualise-geometry-coot report-grade-(L01,L02)
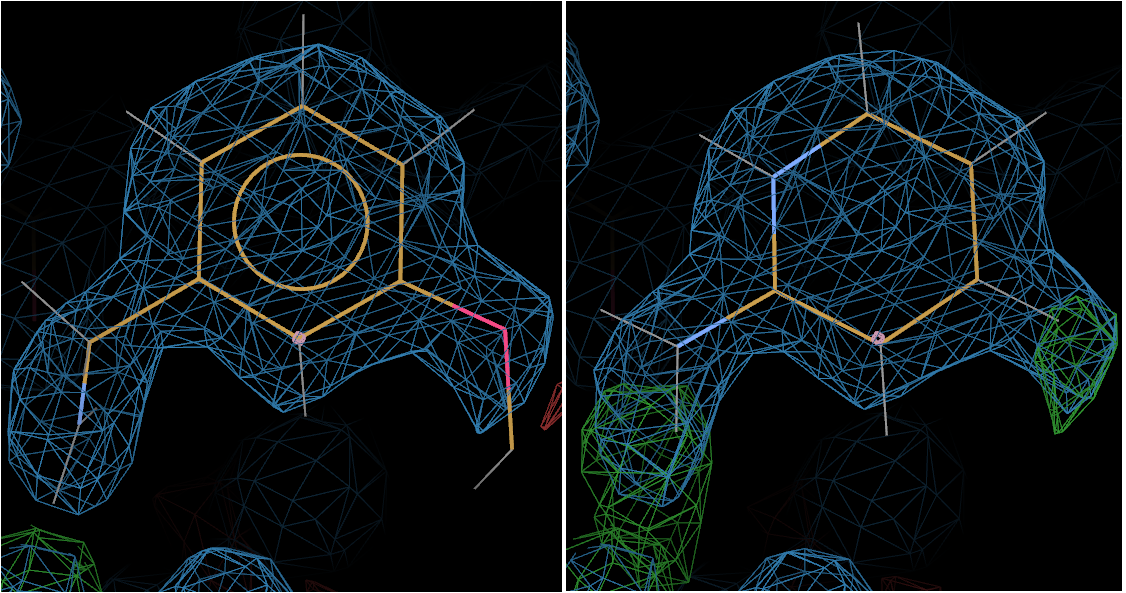
The above picture shows L01 on the left and L02 on the right.
Now the +ve difference density around L02 gives the game away - clearly suggesting that L01 is bound.
And indeed L01 is the ligand shown bound in the deposited structure 3rxt.
Follow on
The above results may suggest, on the face of it, that L01 (ligand code SZ4) and not L02 (ligand code 2AP) is bound.
However, when designing cocktails for use in fragment based x-ray screening, one of the key points is to try to ensure shape diversity between the components of individual cocktails to aid in easy identification of which component may have bound.
In the above example, this is most certainly not the case. In fact L02 is a perfect substructure of L01, making exclusion of L02 as a potential binder impossible from this experiment. A deconvolution experiment - soaking both components individually - would be required.
Indeed, this is exactly what Yamane et al. did, confirming that L02 does indeed bind. They report an IC50 for L01 of 188microM but the binding of L02 was so weak that standard enzyme kinetic studies were insensitive. X-ray crystallography however was able to detect and show binding. 3rxc shows trypsin soaked with L02 and 3rxd is soaked with L01.
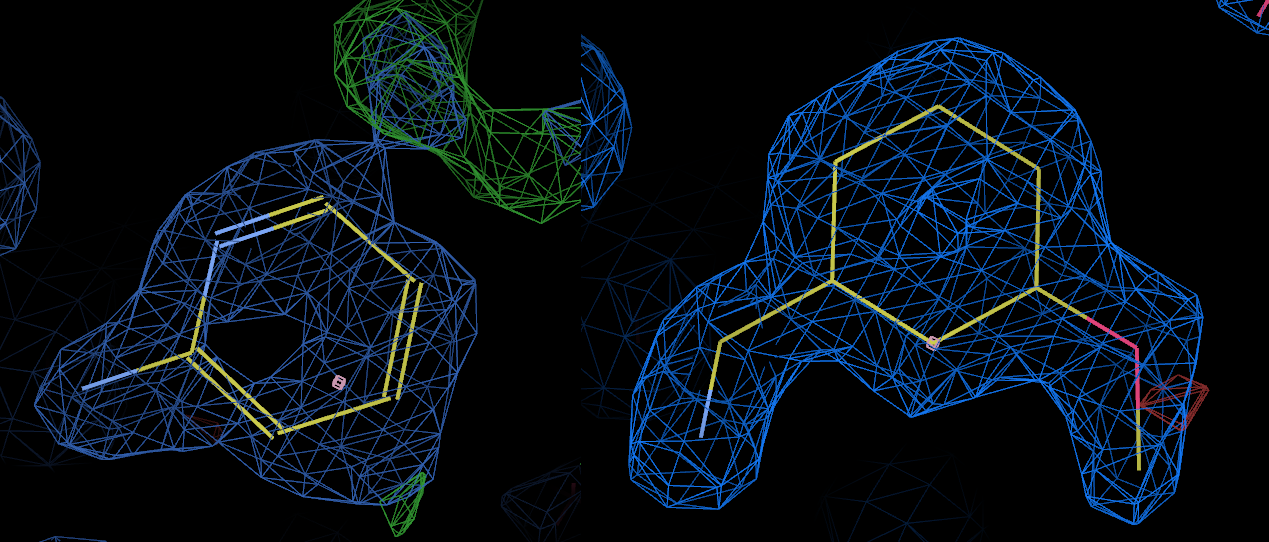
The above picture shows the bound ligand in 3rxc (left) and 3rxd (right) with the 2fofc maps contoured at the same level. Clearly L02 is bound in 3rxc, although the density appears somewhat weaker than the density for L01 in 3rxd.
If the components of the cocktail were truly shape diverse, would it be possible to "deconvolute" their binding modes (without doing a deconvolution experiment) and also potentially tell anything about their relative binding affinities?
Quite possibly - by refining the structure with both components present, making use of BUSTER's occupancy refinement capabilities and showing that the difference density features were diminished relative to refinement of the structures with the individual components.
In this case:
- Take a copy of the final post-refined structure with L01 (postrefine-grade-L01/refine.pdb) and with an editor, add in the coordinates of ligand L02 taken from postrefine-grade-L02/refine.pdb.
- Change the residue number of L02 to 4001
- Assign alternate conformation code A to L01 and code B to L02
- Reset the occupancy of all atoms in both L01 and L02 to 0.5
- Save the modified pdb file (say input.pdb)
- Generate a gelly input file to tell BUSTER to refine the relative occupancies of L01 and L02:
pdb2occ -p input.pdb -o occupancy.gelly
- Now run BUSTER:
refine -p input.pdb -m 3rxt.mtz -Gelly occupancy.gelly -l grade-L01.cif -l grade-L02.cif -d refine > refine.out &
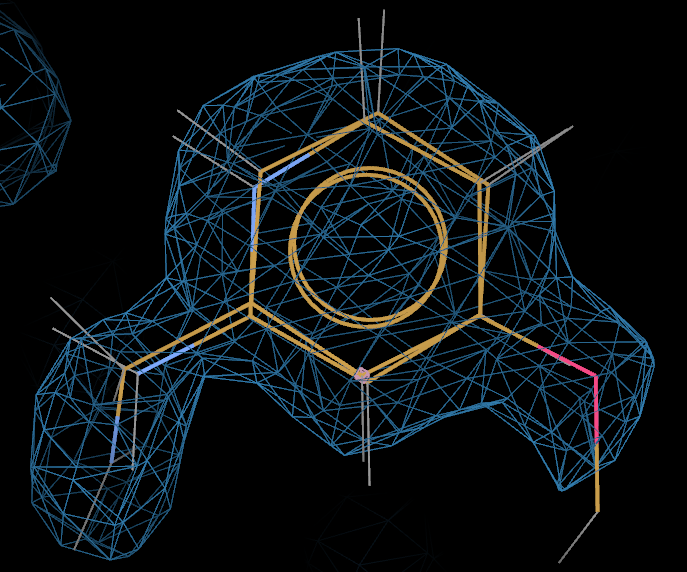
Run buster-report:
buster-report -d refine -dreport report
Look at the output, at the "ligand report" tab
firefox report/index.html
The centre column lists the refined occupancy for both ligands - in this case ~0.79 for L01 and ~0.21 for L02.