Pipedream Tutorial 4
This example illustrates the use of Pipedream where the input are the unprocessed x-ray images, rather than a pre-processed data set.
The example used from released PDB entry 6CBX - "human SET and MYND Domain Containing protein 2 with MTF1497"
The images are available for download at https://data.proteindiffraction.org/sgc/SMYD2_6cbx.tar.bz2
Click on the link to download the images and then unpack and uncompress them.
The reference structure and input model used is from the related PDB structure 6CBY
(A) Restraint dictionary generation
Generate a restraint dictionary for ligand MTF1497 with grade2, again using a smiles
grade2 'Cc1cc2n(cnc2c(N)n1)[C@@H]3CN(C3)C(=O)c4ccn(C[C@H]5CCN(CC5)[C@@H]6CCCCC=C6)c4' -r LIG
(B) Pipedream run
The reference structure 6CBY contains solvent and a ligand that need to be removed prior to use:
egrep -v " EW4 | HOH " 6cby.pdb > input.pdb
We are now ready to run Pipedream as follows:
pipedream -imagedir <dir> -nofreeref -useaniso -apcommands "ReversePhi=yes" -xyzin input.pdb \
-remediate -sidechainrebuild -rhofit LIG.restraints.cif -d pipe &
- Rather than use pre-processed data, as in the previous examples, we are now using the unprocessed images. The use of -imagedir <dir> tells Pipedream where the images are located.
- Note that the use of -imagedir and -hklin are mutually exclusive. You can specify one or the other, but NOT both.
- Note the command -apcommands "ReversePhi=yes" must be specified. The data were collected at APS on 19-ID and the direction of the rotation axis is reversed relative to the standard convention.
- A reference mtz file (1dpw.mtz) containing a freeR set is NOT being used and thus a reference mtz file will be generated from the input model with a new freeR set. The -nofreeref flag MUST be specified to acknowledge this.
- The -useaniso command tells Pipedream to use the Staraniso output mtz file from autoPROC for all subsequent steps.
- There are two copies of the protein in the asymmetric unit. The use of the -chains "A B" option ensures that the limited MR step treats each chain independently. Likewise the -xclusters 2 option tells Rhofit to look for and fit the ligand into the top 2 sites.
- The use of the -remediate -sidechainrebuild options tell Pipedream to run PDB_REDO program sideaide as part of the refinement protocol to both check and correct sidechain rotamers and also to complete incomplete / stubbed sidechains.
(C) Analysis
The main output file summary.out shows that the job has run to completion and without any errors.
- The second main section indicates that the input data was an unprocessed set of images and that it has been successfully processed with autoPROC. The salient processing statistics are tabulated. For a more detailed summary of the data processing, look at the autoPROC report file summary.html.
- Note that autoPROC has been run having been told to index the data with the cell dimensions and space group read from the reference structure. If autoPROC is unable to index data in the specified cell / space group then Pipedream will terminate with an appropriate message.
- The rest of the file indicates that the structure has seemingly been well refined, Rhofit has found a good solution (for each protein chain) and it has been post-refined well.
- The final statistics (R = 18.1, Rfree = 21.2) are a slight improvement than those for the 6CBX structure as deposited (R = 18.1, Rfree = 22.3), coupled with an improvement in resolution of nearly 0.3 Angstrom .
- Look at the output from buster-report:
firefox report-grade-LIG/index.html
- Look at the overall statistics listed on the main tab and also look at the "Ligand report" tab. There is nothing there of any concern. The refinement statistics look very good and the ligand geometry looks good too.
- Now look at the "Molprobity analysis" tab. For the most part, this looks good. Some manual inspection and intervention may be warranted. Again, Pipedream is NOT designed to give a "highly polished", deposition ready structure.
More importantly, is the ligand solution correct? Pipedream will only select what Rhofit classifies as its "best" solution for post-refinement.
So, lets look directly at the Rhofit output:
visualise-rhofit-coot rhofit-grade-LIG
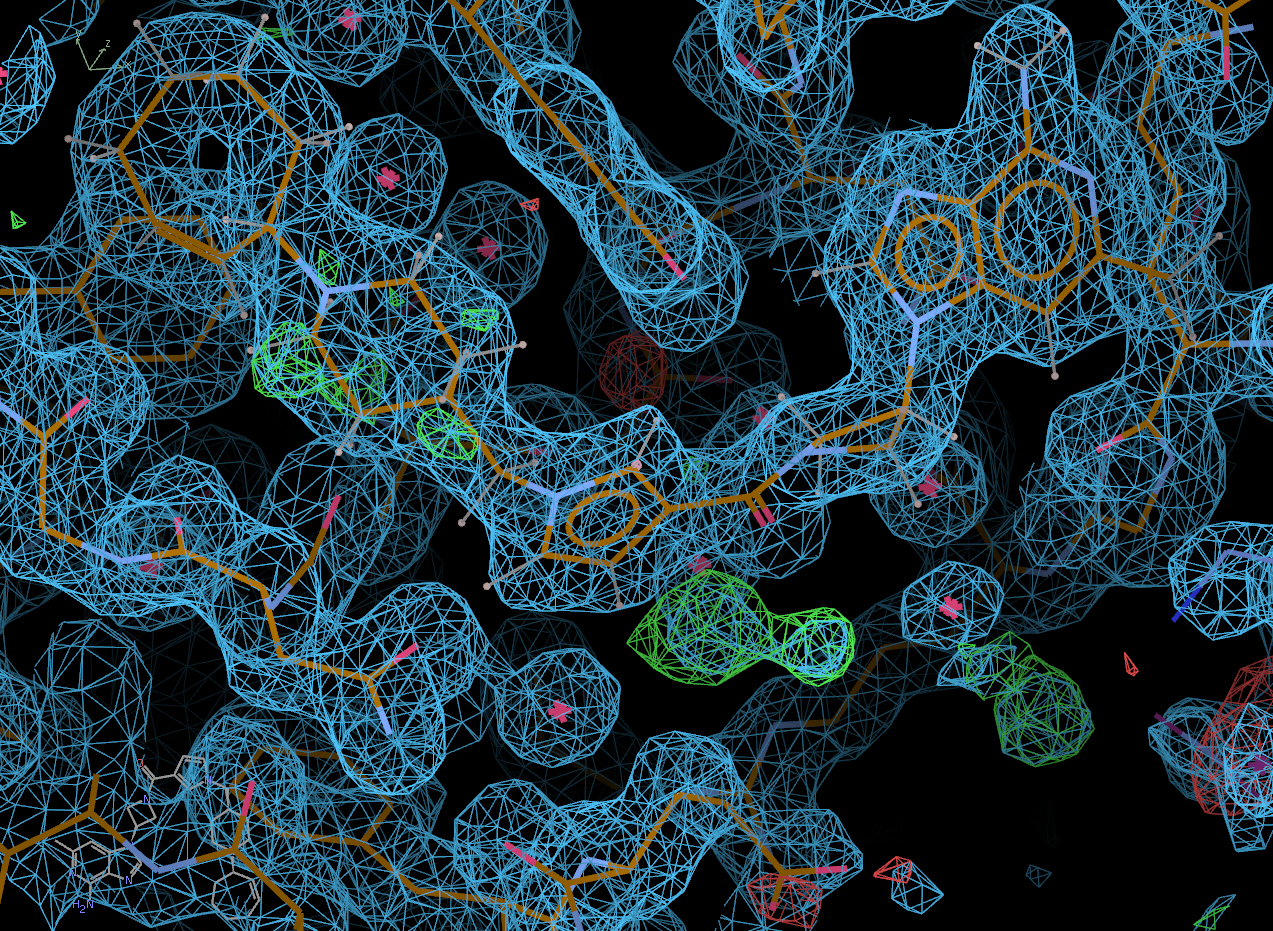
Coot will open, initially displaying its "best" solution and the difference density map.
In the pop-up window, select the "Protein visible" button to also show the protein and the 2fofc density.
Click on the "Next position" tab to scroll through all of the potential solutions that Rhofit built. Solution 0 appears to be correct.
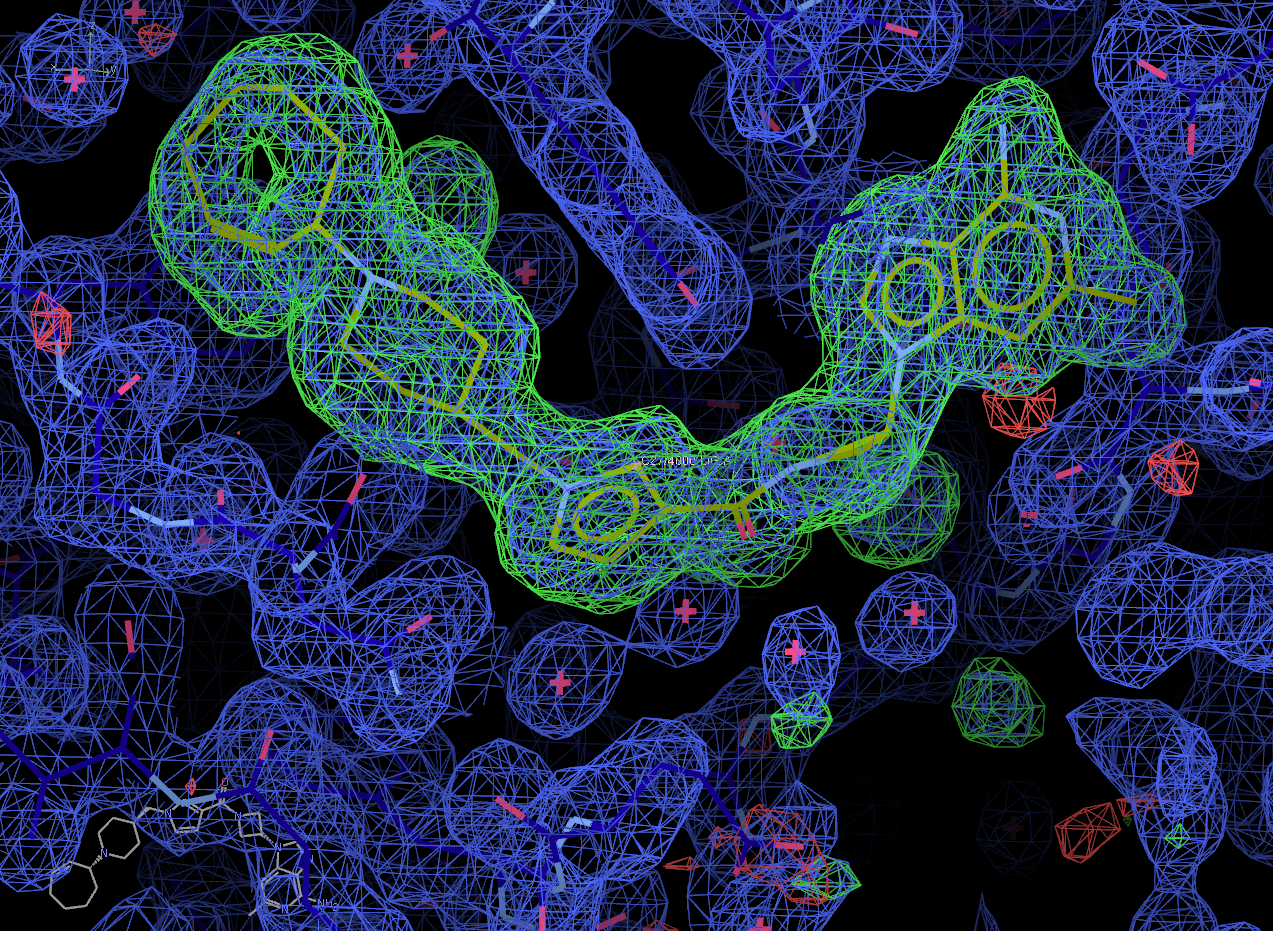
Now select the "Next search region" tab to look at the second binding site. Again, solution 0 appears to be correct.
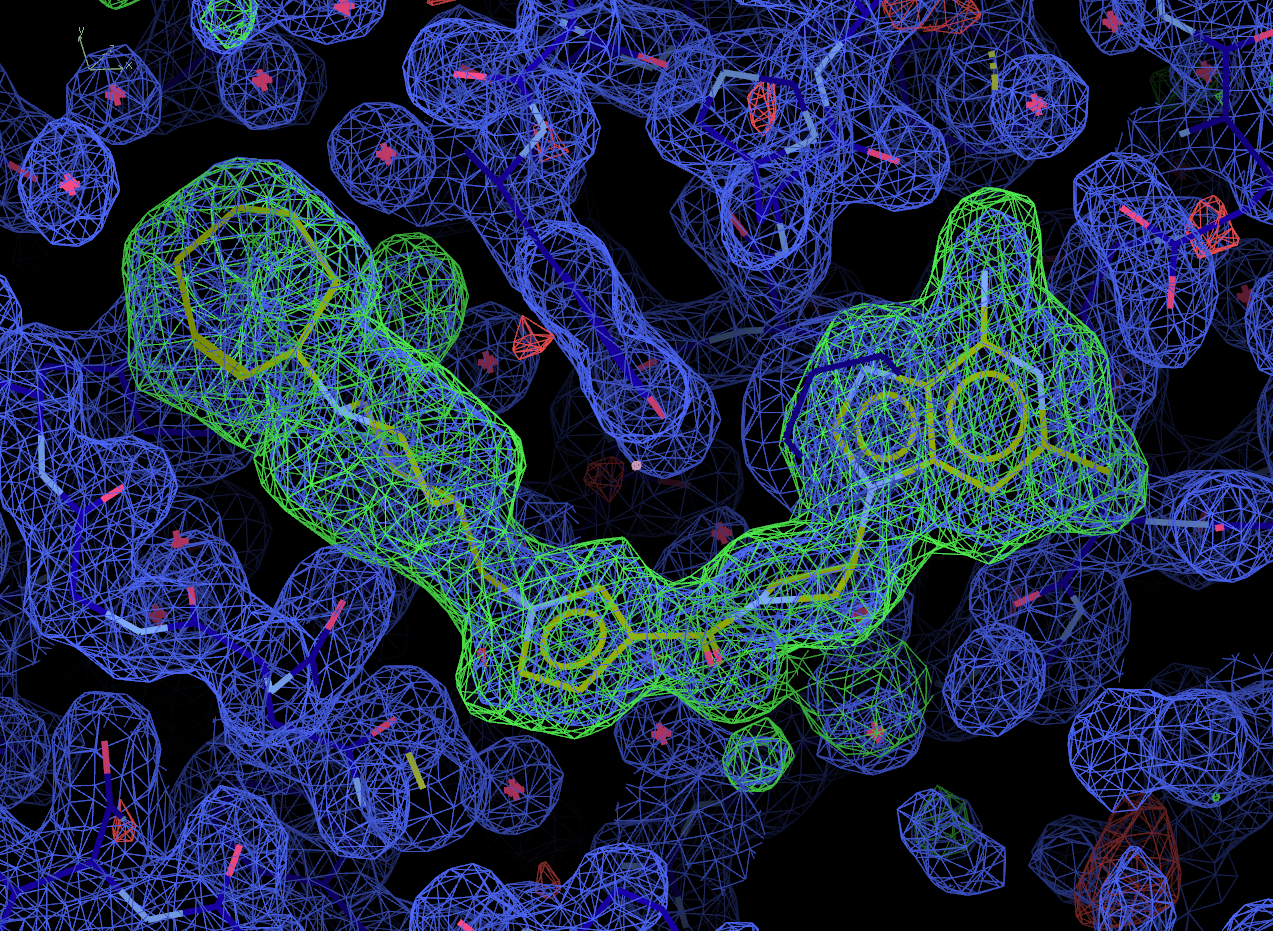
Look at the final post-refined structure and map using visualise-geometry-coot:
visualise-geometry-coot postrefine-grade-LIG
chain A:
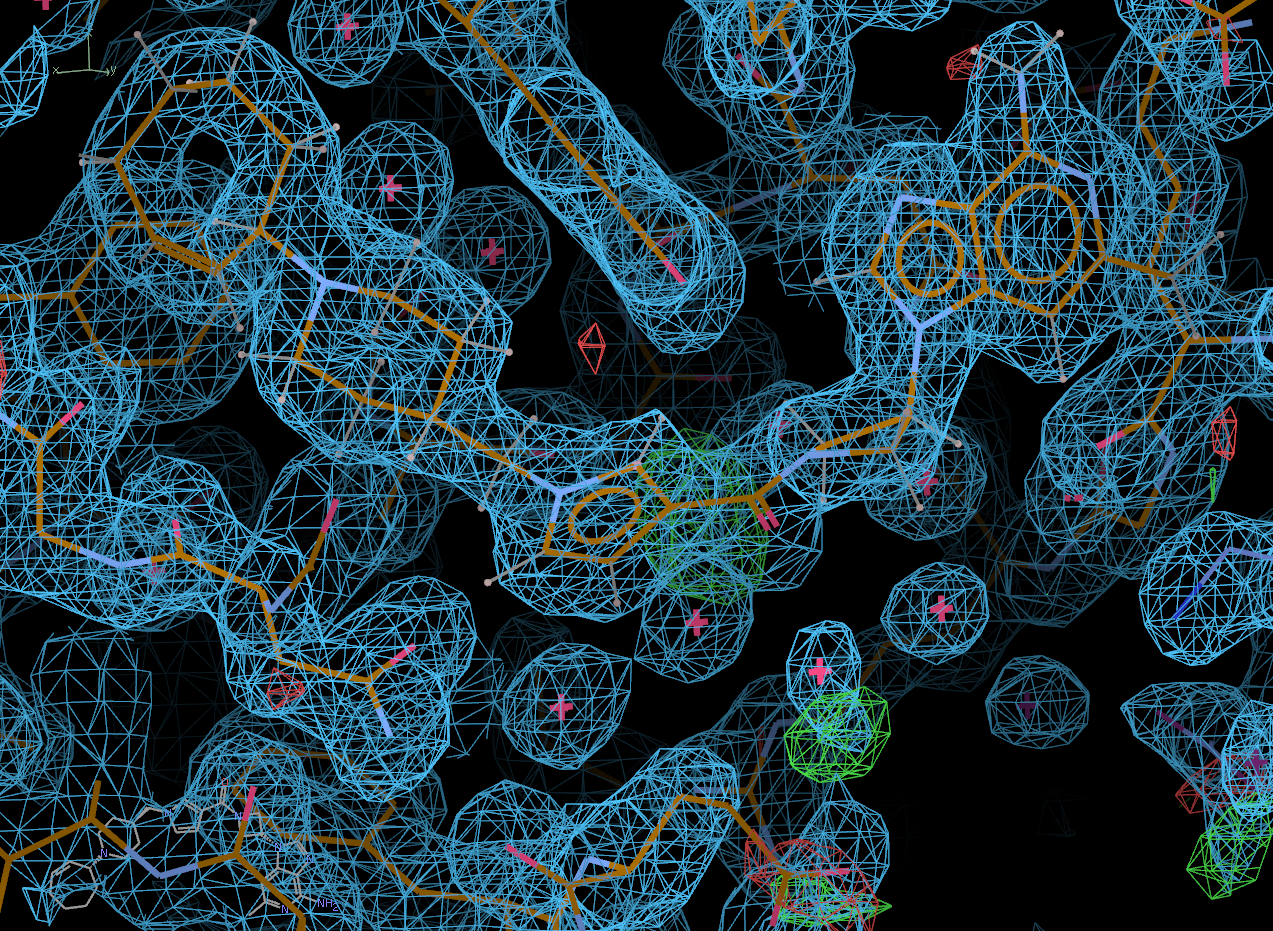
chain B: