| Attachments | |
|---|---|
| nodomain-summary.out | 5K |
| 1w50.pdb | 282K |
| 4ke1.mtz | 1MB |
| nodomain-residues.list | 1K |
| 4j0p.pdb | 512K |
| rigid.dat | 104B |
| domain-residues.list | 69B |
| hinge.png | 139K |
| hinge-result.png | 214K |
| 4dh6-hinge.pdb | 229K |
| domain-summary.out | 7K |
(revision 1.0, February 2015)
| Note | The features described in this tutorial require Pipedream version 1.1.0 or later. |
|---|
This tutorial follows on from Tutorial 5 to illustrate the use of Pipedream with multiple input models, specifically where domain movement, such as hinge bending, is present. This tutorial uses the same beta-secretase structures as in Tutorial 5. The experimental data is taken from PDB id: 4KE1. The three input models used are PDB id's: 1W50, 4DH6 and 4J0P.
| 4ke1.mtz | the structure factors as supplied by the pdb for 4ke1, converted to mtz format |
| 1w50.pdb | the model coordinates as supplied by the pdb for 1w50 |
| 4dh6-hinge.pdb | a modified set of model coordinates from PDB entry 4dh6 |
| 4j0p.pdb | the model coordinates as supplied by the pdb for 4j0p |
What happens if your target structure exhibits more significant domain movements, such as hinge bending, as well as localised loop rearrangements?
Pipedream bases its determination of which residues differ and then comparison of which model fits best on an analysis of pairwise RMS deviations between residues. However, whole domain movements, such as hinge-bending, will compromise this analysis and lead to incorrect outcomes.
Therefore, such domain movements must be factored out before structure comparison is carried out.
Luckily, this is catered for in Pipedream, through compulsory rigid body refinement at the start of the initial refinement step and the ability to give Pipedream a specific rigid body definition file to "divide" the structure into domains, which are refined independently.
This tutorial uses the same input files that were used in Tutorial 5, with one exception.
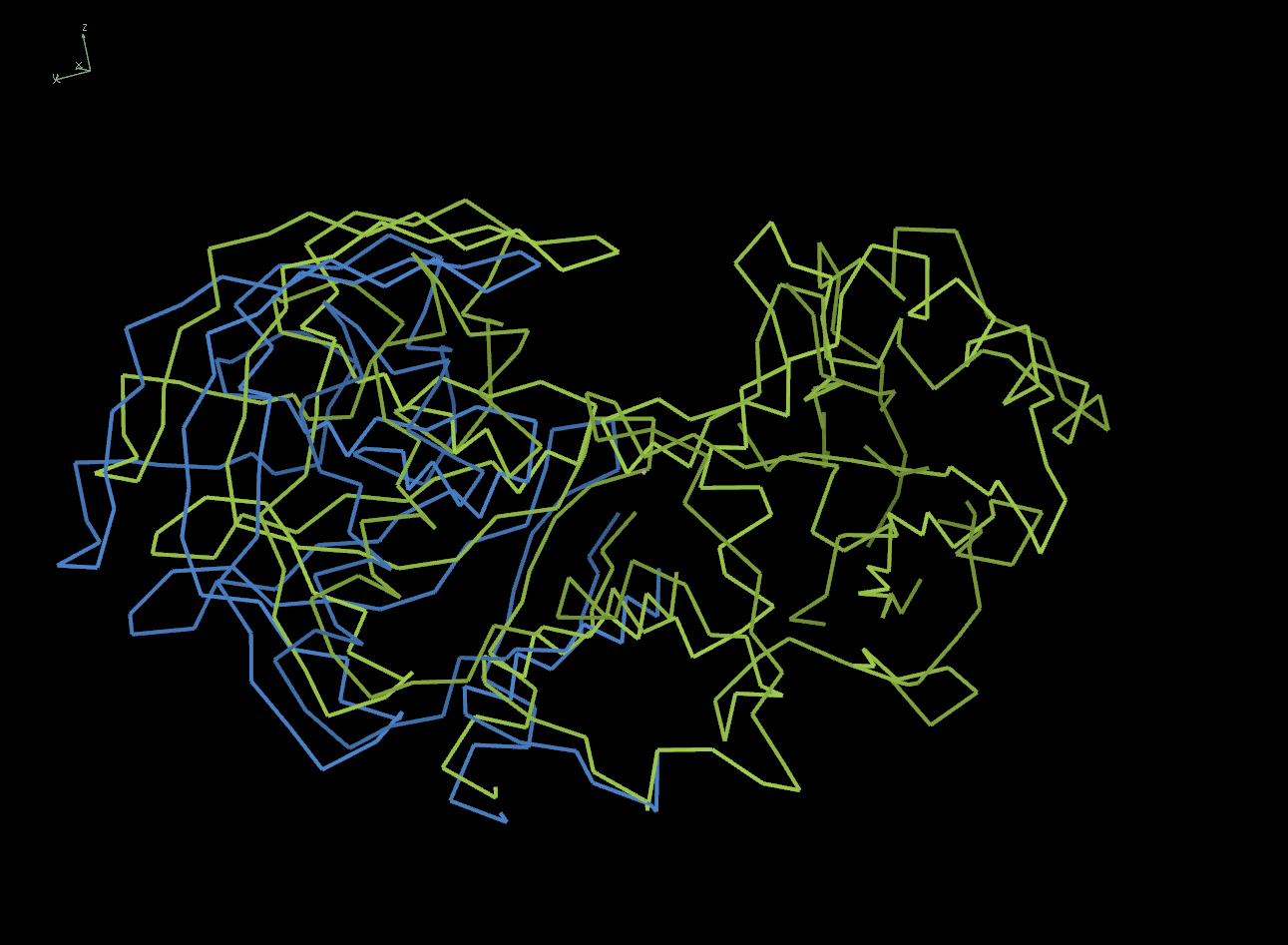
The 4dh6 structure has been altered in Coot to introduce a random rotation of its n-terminal domain (residues -1 to 183).
The image below illustrating the 4dh6 structure (yellow) and the modified model (blue), showing the rotation introduced into the n-terminal domain.
Prior to running Pipedream, we also need a rigid body definition file. In this case, there are just 2 domains:
n-terminal domain: -1 to 183 c-terminal domain: 184 - 385
See the main BUSTER program documentation for a description of the format of the rigid body definition file.
The file that we need is here. Note the first line of this file. This is setting explicit resolution limits for the rigid body step in BUSTER. We are limiting the high resolution limit to 4.0A as it will increase the radius of convergence for rigid body refinement.
(C) Pipedream run and Analysis
We are now ready to run Pipedream.
First, lets see what will happen if we do not give Pipedream the rigid body definition and let Pipedream treat the whole structure as a single rigid body:
pipedream -hklin 4ke1/4ke1.mtz -nofreeref -xyzin 1w50.pdb,4dh6-hinge.pdb,4j0p.pdb -d nodomain
See the main output - summary.out
We know from the results of Tutorial 5, that we are expecting that 4dh6 will be the best match to the experimental data.
But look at the output file. The mean Z-score is significantly worse than either of the other two models. In fact, Pipedream has selected 1w50 as the best match. 1w50 is the wrong model, especially as the loop described in Tutorial 5 is occluding the binding site.
Look at the long list residues that pipedream selected as different between the models - here. These residues make up most of the n-terminal domain,
So, now lets run Pipedream with the rigid body definition file:
pipedream -hklin 4ke1/4ke1.mtz -nofreeref -xyzin 1w50.pdb,4dh6-hinge.pdb,4j0p.pdb -rigid rigid.dat \ -rhofit 1R6.restraints.cif -rhothorough -d domain
See the main output file - summary.out
The result now is very different. The mean Z-score for 4dh6 is now significantly higher than from the other two. Pipedream has now selected the expected model, 4dh6. You will notice that the final result is the same as in Tutorial 5.
Look at the list of residues selected now for analysis - here. This list matches the result for Tutorial 5 and is made up mostly of the residues in the loop that moves in/out of the bonding site.
The image below now shows the final refined structure (orange), the input 4dh6 model (blue) and the deposited 3ke1 structure (green), showing that the domain shift has been fully corrected by the rigid body refinement.
