rhofit test on Wlodek et al. (2006) test set.
Content:
- introduction and methods
- rhofit results
- comparison between rhofit and FitMAP results
- autobuster refinements from rhofit best fit ligands
- test of rhofit -scanchirals: can rhofit work from ligand with scrambled chiral centers
1. introduction and methods
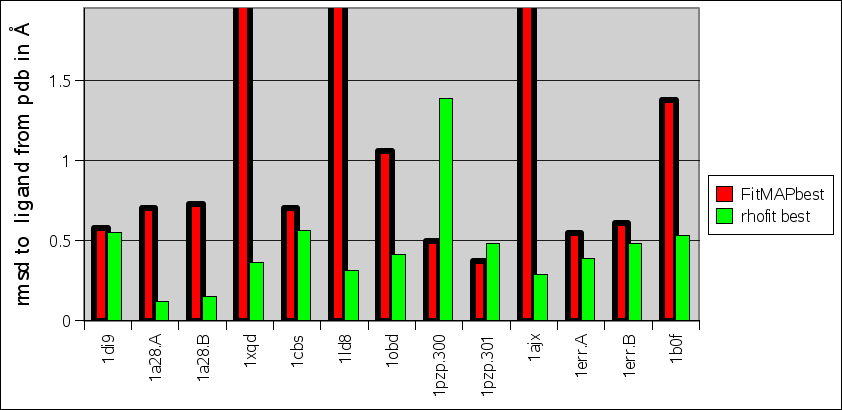
rhofit is designed to flexibly fit ligand molecules into difference density. The AFITT program by OpenEye Scientific Software has similar capabilities. In the paper describing the method Wlodek et al. (2006) Acta Cryst. D 62:741-749, 11 protein-ligand complexes are used as a test of the procedure (see Tables 1 and 2). It is found that the procedure can refit ligands into the 2Fo-Fc density map from the model refined with the ligand with a maximum rmsd to the pdb position of 0.8Å.
To test that BUSTER with rhofit can cope with these ligands it was decided to apply the procedures on the Wlodek et al. (2006) test set. Rather than directly using the 2Fo-Fc density map from the refined model the following procedure was adopted:
- The relevant ligand was striped out from the pdb entry.
- The protein was then refined with autoBUSTER -L. This results in water insertion being applied during refinement. Before the final cycle likely areas for ligand binding are found and the model water molecules in this area are removed from the model, but kept for the purposes of defining the bulk solvent mask. This is found to improve difference density.
- Model ligand coordinates were obtained from the PDBeChem service or derived from SMILES strings using the cactus online SMILES translator service. In both cases this results in models being obtained from the CORINA program.
- The model ligand was then placed into density using rhofit and FitMAP. In cases with more than one ligand site rhofit was asked to identify the known number of sites.
2. rhofit results
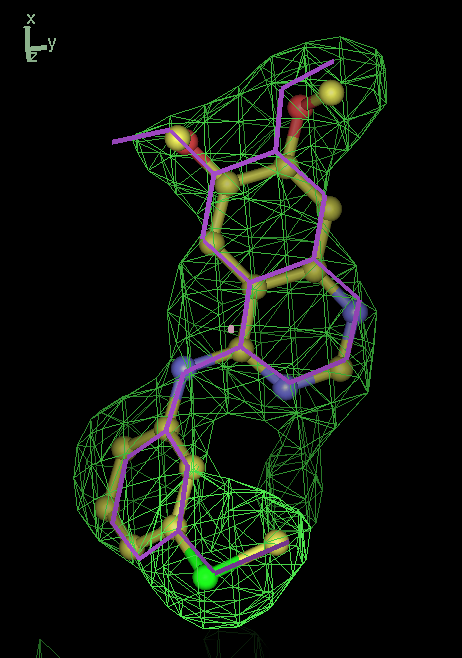
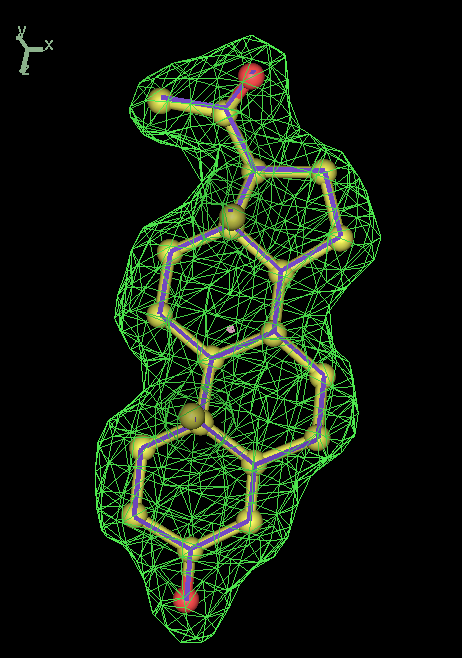
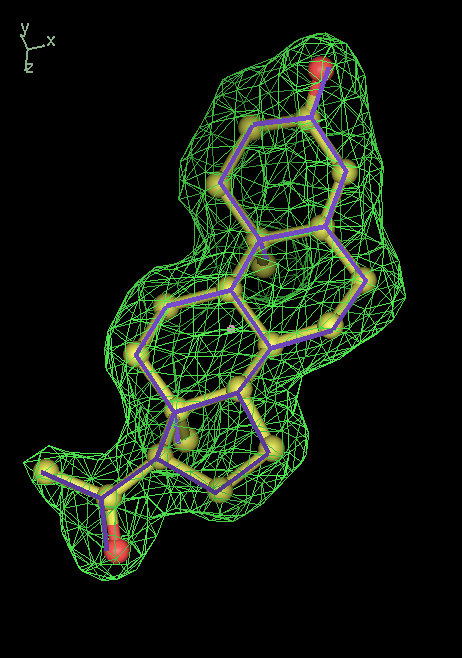
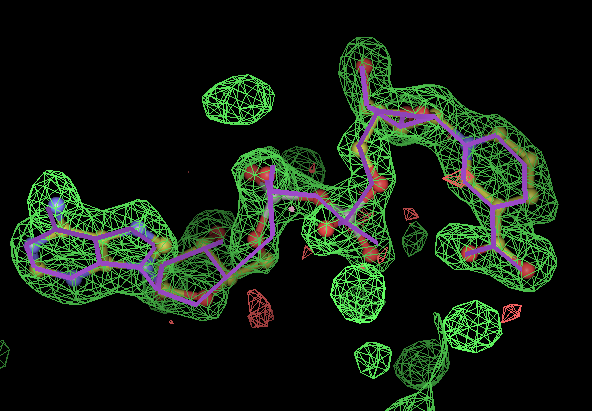
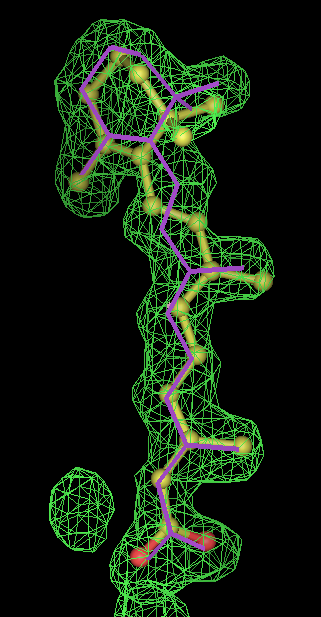
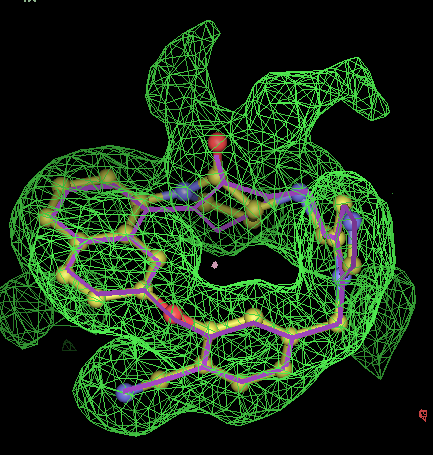
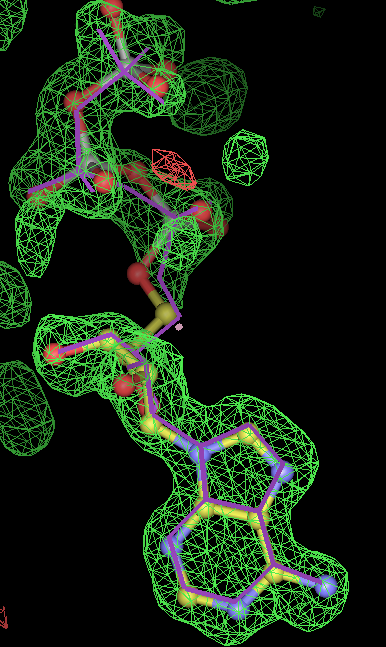
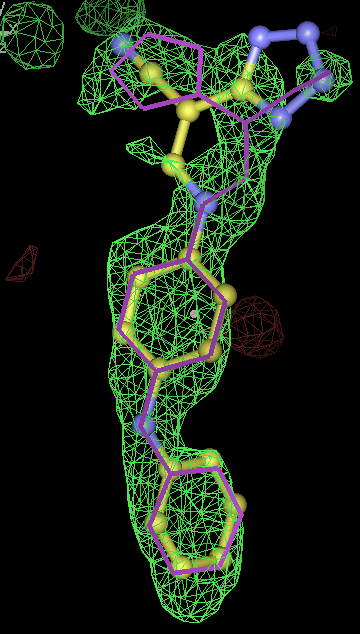
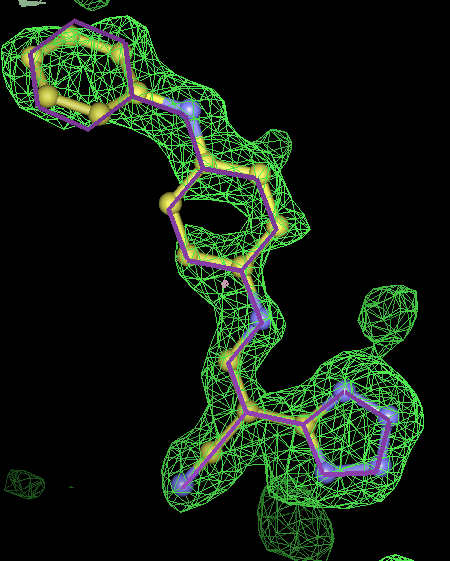
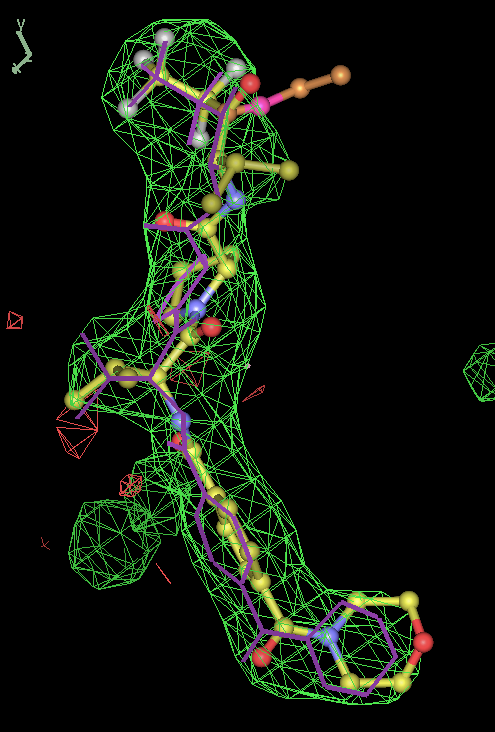
- Click on the picture to expand (or right click to get in another tab)! Difference density as found by autoBUSTER refine -L contoured at 3.5 sigma in green, rhofit best solution in atom-colored ball and stick. pdb ligand position in purple lines.
- In this case only autoBUSTER refine -L failed to identify this weak binding site. An autoBUSTER refine -Lpdb was then used with the binding site being manually defined by the waters inserted into it in the original run.
- The SEI ligand is covalently bound to the elastase protein via and ester bond to the active site serine residue. This version of rhofit does not explicitly consider covalent ligand protein bonds. Because of this the ligand was considered as unbound with the serine CB and OG atoms being removed from the fixed protein model in the rhofit run. The rhofit best solution places the ligand with atom C71 adjacent (1.0Å) to the active site serine 195 OG atom to which it is bonded. In the picture of the solution the serine side chain is shown in orange and pink. It can be noted that the geometry of the link would be improved by sensible restraints, but the same is true of 1b0f.pdb!
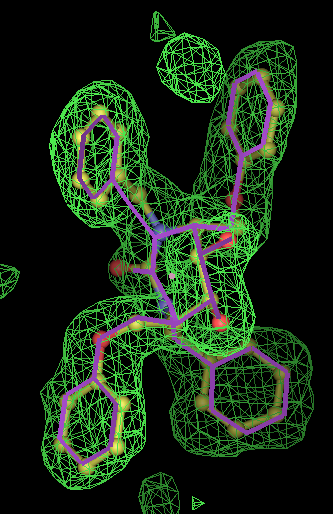
- The PVH "ligand" is a histidine residue covalently bound to the C terminus of the protein, with its carboxylic acid esterified. There are 3 NCS copies and at 3.2Å resolution the density for this is a blob. It is simply not sensible to model this residue without taking into account the its bonds.
3. comparison between rhofit and FitMAP results
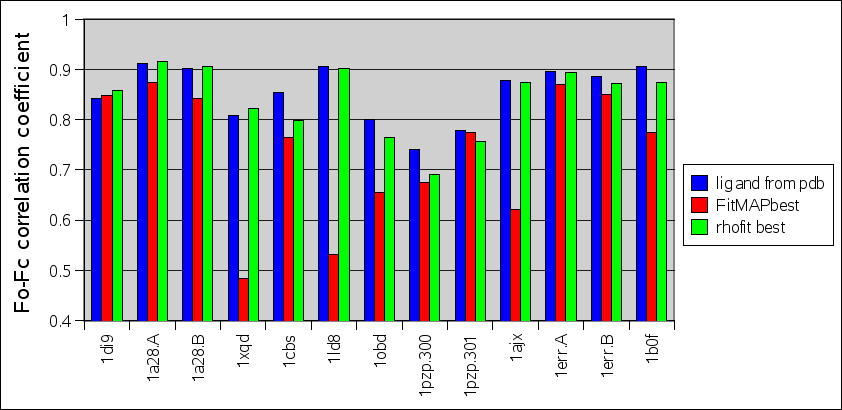
Comparison between rhofit to FitMAP is shown in the charts:
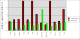
- It can seen rhofit obtains a solution with 0.8Å rmsd of the pdb in 12 out of the 13 cases. In comparison FitMAP gets a close solution in 8 of the 13. In the three cases FitMAP produces poor solutions: 1xqd, 1ld8 and 1ajx. 1xqd is a complex dinucleotide. 1ld8 and 1ajx contain macrocycles and as FitMAP was not designed to cope with such cases.
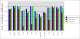
- It can be seen that rhofit consistently produces solutions with a higher difference map correlation coefficient that FitMAP.
4. autobuster refinements from rhofit best fit ligands
to be written up
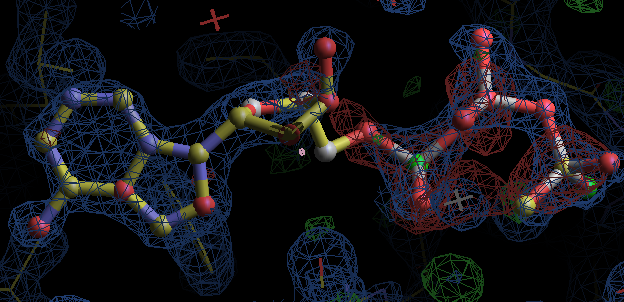
- but most interesting result is for 1obd with ATP. After refinement the density appears to indicate an adenine with a separate partially occupied polyphosphate magnesium ion complex. So this is not an ideal example for fitting! (The EDS map for 1obd shows differences on the MG2++ and phosphate P atoms at 3.5 to 5 sigma.)
5. Test of rhofit -scanchirals: can rhofit work from ligand with scrambled chiral centers?
- In order to test the test the -scanchirals option it was decided to see whether rhofit could obtain the "correct" binding position starting from an ideal ligand model where the chiral centers were delibrately set wrong. The procedure adopted was:
- The SMILES string for the pdb ligand from was got from ligand expo and then flip the 1st, 3rd, 5th, and 7th chiralities in the string. This results in at least half the chiral centres being wrong. (It was decided that this was a more demanding test that flipping all centres which is equivalent to a simple mirror inversion results).
- Ideal (CORINA) coordinates for the scrambled-chirality SMILES string were obtained from the cactus server: http://cactus.nci.nih.gov/translate/
- rhofit -scanchirals was used to fit the density/protein model as described in section 1
- results:
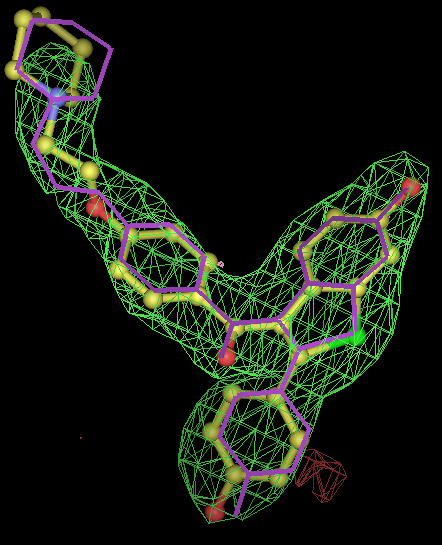
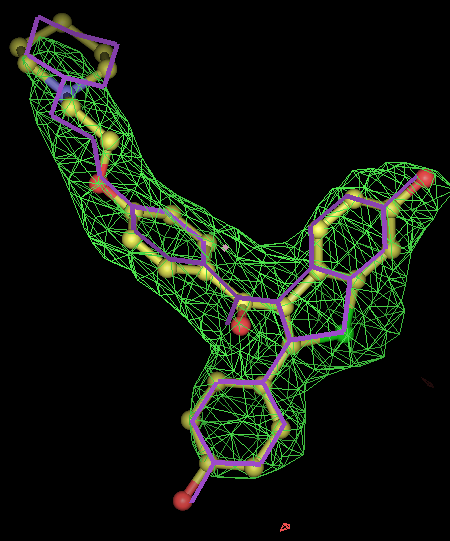
- for 8 chiral centres a divide and conquer strategy was used where the correct chiralities were determined for the two sugar rings were determined separately.
- chiral center is pretty much flat in pdb. From the -scanchiral runs I think it likely that msdchem has the incorrect chirality from the center.
- conclusions: rhofit -scanchirals can identify the correct chiral combination for all of these test cases. Note that this is also a test of how good the density is. At low resolution it is unlikely that a map would have enough information for -scanchirals