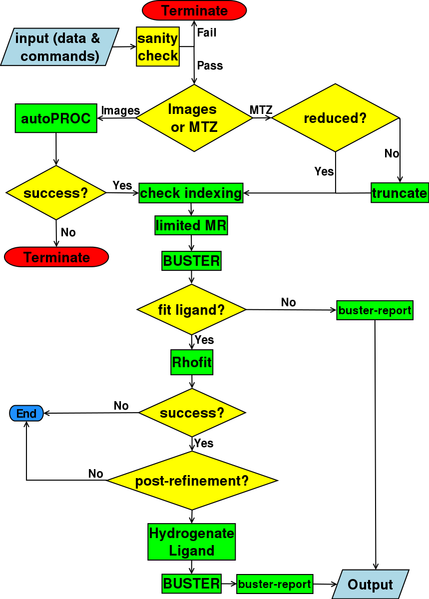
Program Flow
Pipedream links Global Phasing's main packages in a stepwise and essentially linear workflow:
- Data processing (if required) with autoPROC
- Limited molecular replacement (to deal with crystal non-isomorphism / conformational change) using phaser
- Structure refinement using BUSTER
- Automated ligand fitting with Rhofit
- Post-refinement of ligand fit structure, with BUSTER
- Analysis of final structure with buster-report
Input requirements and limitations
The scenario for which Pipedream has been designed, typical for a fragment screening regime, is that you have lots of crystals of your particular target protein from which you will collect x-ray data and solve the structures, both "apo" and soaked with potential ligands.
The main assumption, implicit from the above scenario, made by Pipedream is that the only significant difference between the structures in these experiments will be the presence/absence and nature of a bound ligand. Critically, Pipedream imposes and enforces a requirement that the space group of the experimental data must be the same as the reference structure, as do the cell dimensions (within limits due to inevitable crystal variability and non-isomorphism).
The required input data for pipedream are:
- The experimental data for the soaked crystal. This can either take the form of the unprocessed images, in which case Pipedream with run autoPROC to process the data, or an mtz file if the data have already been processed.
- The structure of the "reference" model. Usually this will be the apo/native structure, but it could be a ligand bound model with the ligand removed from the coordinate file. Optionally, but RECOMMENDED, the mtz file corresponding to the reference model. This way, the same Free R set present in the reference mtz file can be copied over to the experimental data
- Restraint dictionary for the ligand(s) that has(ve) been soaked into the experimental structure.
Output
Pipedream is designed to be run in the background or submitted through a batch queue system. As such, it does not write anything to standard output. ALL of the output from Pipedream and from the separate packages that it calls is written into a new, clean directory, which MUST be specified on the command line.
All output is written in the defined (root) directory as follows:

In addition, Pipedream writes its own information and also collates, summarizes and tabulates the main information from each of the individual stages in the file summary.out, which should be the first file to look at.