Using hydrogens in high-resolution structures with autoBUSTER
The version of autoBUSTER released in February 2011 has support for refining protein models in which every hydrogen atom is modelled and contributes both to the X-ray and to the geometry term. The positions of the hydrogen atoms are restrained in our standard amino-acid dictionary by bond-length, bond-angle, torsion, planarity and ideal-contact terms.
We have run refinements with full hydrogenation on several dozen proteins and find that, at high and moderately high resolutions (1.8A or better), there is a pronounced improvement in the free R value achieved by using our hydrogenate tool (which uses the reduce tool from molprobity) to add all hydrogens explicitly and to perform HQN flips, and then refining taking advantage of these hydrogens.
| Resolution bin | Improvement in free R | cases tested |
| 1.0A | 0.0089 ± 0.0022 | 17 |
| 1.2A | 0.0105 ± 0.0042 | 21 |
| 1.5 | 0.0096 ± 0.0039 | 23 |
| 1.75 | 0.0078 ± 0.0040 | 21 |
| 2.0 | 0.0001 ± 0.0029 | 19 |
Note that in these tests we used a fixed X-ray weight for the comparison, since the effects of changes in the X-ray weight can add a lot of noise to the statistics. This would be a laborious protocol to apply for individual refinements, and in particular if you have been refining a structure through several rounds of BUSTER the X-ray weight should have been automatically set; so don't worry about this for your refinements.
A worked example
Let's consider the structure 3a4r; this is a 1.0A structure of a ubiquitin-like domain, deposited in September 2009. 3a4r.pdb and 3a4r.mtz are attached.
Ensure that the reduce program is on your path, and run
hydrogenate -p 3a4r/3a4r.pdb -o 3a4r-fullH.pdb
This will provide output
hydrogenate uses the 'reduce' tool; if you publish work which uses it, please cite Word, et. al. (1999) J. Mol. Biol. 285, 1735-1747. For more information see http://kinemage.biochem.duke.edu Residues EDO SO4 not hydrogenated; give dictionaries (with the -l dic1 dic2 ... option) to do this
SO4 is not hydrogenated under physiological conditions; as for EDO, you can generate a grade dictionary (assuming you have the prerequisite software installed) with
grade_PDB_ligand EDO
or download EDO.grade_PDB_ligand.cif
and rerun
hydrogenate -p 3a4r/3a4r.pdb -o 3a4r-allH.pdb -l EDO.grade_PDB_ligand.cif
Load 3a4r-allH.pdb into coot and have a look around. Note that reduce has placed hydrogen atoms to make hydrogen bonds where this is possible, and has flipped some ASN, HIS and GLN residues - for example, residue A352 has been flipped round to allow one of the hydrogens on the nitrogen to make a good hydrogen bond to the water A232.
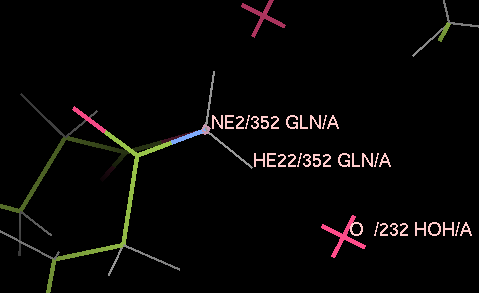
The refinement
Now start a refinement with
refine -p 3a4r-allH.pdb -m 3a4r.mtz -autoncs -l EDO.grade_PDB_ligand.cif \ -d allH | tee allH.lis
Note that we need to pass the hydrogenated ligand dictionary to refine. The refinement should be uneventful, and conclude with
best refinement in BUSTER-GELLY-TNT reached for FP,SIGFP with R/Rfree 0.1982/0.2120
If you had run a refinement of the original unhydrogenated structure, it concludes with
best refinement in BUSTER-GELLY-TNT reached for FP,SIGFP with R/Rfree 0.2016/0.2130
What does molprobity think?
| molprobity term | Deposited 3a4r | Refinement without H | Refinement with H |
| All-atom clash score | 7.48 (42%) | 7.48 (42%) | 4.15 (81%) |
| Bad rotamers | 2/131 | 2/131 | 2/131 |
| Molprobity score | 1.55 (63%) | 1.55 (63%) | 1.34 (83%) |
The molprobity clash score includes a substantial contribution from considering close contacts involving hydrogen atoms; refinement with explicit hydrogen atoms which are allowed to move in a chemically-plausible fashion to relieve close contacts will lead to much lower values of the clash score. Indeed, in some of the highest-quality structures, we have seen cases of manual rebuilding and refinement with explicit hydrogen atoms leading to a clash-score of zero.
A feature revealed by this refinement
As always, load the result of the refinement into coot with
visualise-geometry-coot allH
It should look good, with the almost-separated atoms characteristic of data at 1A resolution. But the 'geometry issues' panel
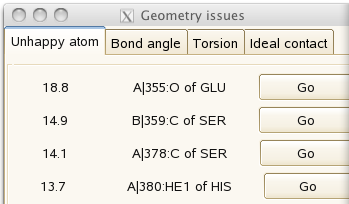
gives a couple of unhappy atoms. If you click on 'Go', you can see that the side-chain of histidine A380 needs to be flipped to make a hydrogen bond with the main-chain carbonyl of A355:
| before | after |
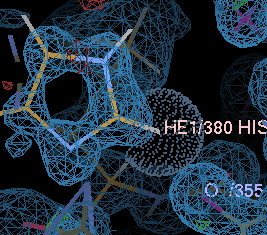 |
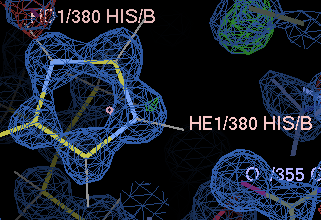 |
molprobity has sophisticated algorithms for picking flips, but unfortunately isn't aware of close contacts across crystal contacts.