autoBUSTER Example Application: RNA refinement and restraints to favor canonical puckers
Background
- This page presents recent work on improving geometry restraints for RNA and on how to use utility restraints to favor canonical pucker angles.
- As an example we will use the structure 3g4m "Crystal structure of guanine riboswitch bound to 2-aminopurine". This is a 2.4Å resolution structure that was refined with CNS. We will refine this structure using BUSTER and see whether it is possible to improve its geometry.
- Three useful validation/analysis tools will be used
Starting Files
Control Refinement
- Refining with BUSTER with currently distributed restraints:
refine \
-p 3g4m.pdb \
-m 3g4m.mtz \
-d 01_control \
-l NCO_correct.cif \
-l 2BP.grade_PDB_ligand.cif \
-M TLSbasic -nbig 10 \
AnalyseVoidsLast="no" > 01_control.log &
- notice we use the TLSbasic macro for TLS refinement and turn off the hydrophobic void correction normally done in the last cycle.
- The result of the refinement is:
- So BUSTER refinement results in an increase in Rfree and the Rfree-Rwork gap together with the degradation of both MolProbity and coot validation scores. This is not good.
- The bond angles are of particular concern. BUSTER automatically adjusts the X-ray weight to achieve a final RMS bond deviation from ideal of 0.010Å. For protein refinement this normally results in an RMS angle deviation below 1.0 degrees. In this refinement the deviation is approximately twice as bad. This is then reflected in the dire molprobity residues with bad angles metric.
Improved Nucleic Acid Restraints
- The poor bond angle deviation metric found in the control refinement is caused by the currently distributed nucleic acid restraint dictionary file using unrealistic large values for the "sigma" of all bond angle restraints. This has been corrected in the new restraint dictionary that can be downloaded above.
- The new dictionary also revises the restraints used for torsion angle restraints removing some unrealistic terms. See file header for details.
- The new dictionary will replace the old in the next release of BUSTER.
- To use the new dictionary use the !autoBUSTER refine command line option TntDictionary_nuclgeo with the full path of the file. (Here we use the pwd command to supply the current directory).
refine -p 3g4m.pdb \
-m 3g4m.mtz \
-d 02_better \
-l NCO_correct.cif -l 2BP.grade_PDB_ligand.cif \
-M TLSbasic -nbig 10 AnalyseVoidsLast="no" \
TntDictionary_nuclgeo=`pwd`"/nuclgeo_new_wiki_2011.dat" \
> 02_better.log
- Refinement with the new dictionary results in a marked improvement, over results with the old.
- Overall Rfree is improved by 0.5% compared to before. The gap also remains better and there is a marked improvement in molprobity scores
- Compared to the pdb there is a small increase in Rfree but this could be to due to more complete refinement. Some geometry scores are slightly worse but the clashscore is markedly better.
- Importantly after refinement the model can be improved.
- use the visualise-geometry-coot tool to visualise which part of the 02_better model have the worst geometry.
- Doing this shows that the new torsion restraints indicate that there is a very bad torsion violator:
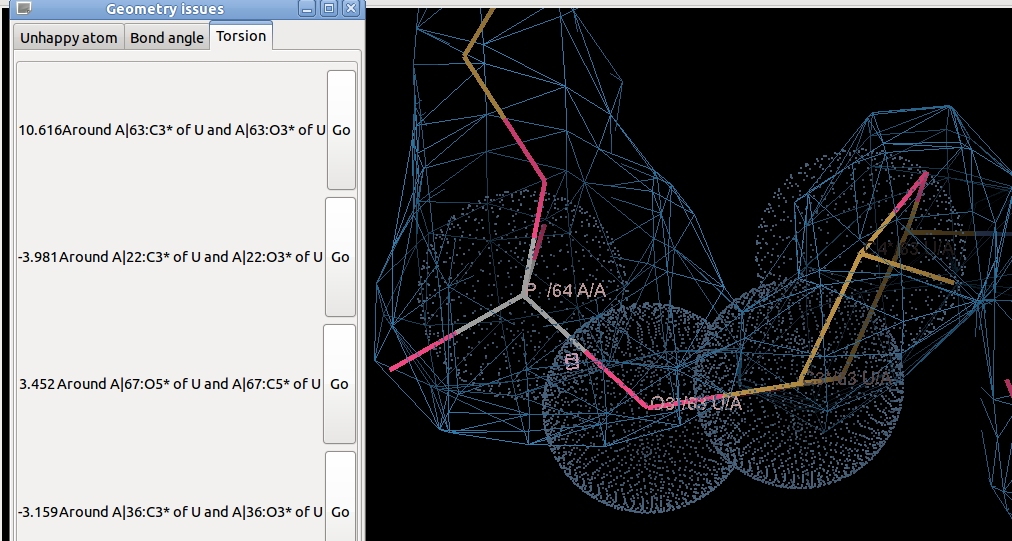
- The torsion angle C4' U63 = C3' U63 = O3' U63 = P A64 is at 54 degrees. This torsion is the "epsilon" torsion and a positive value is most unfavorable - best shown figure in figure 4-25 Saenger (1984) "Principles of Nucleic Acid Structure". The atom O3' is at the edge of the density.
- Residue 63 also has bad bond angles. MolProbity lists it as having unfavourable pucker, angles and conformer.
- Modeling the structure in coot (after using the pdbvconv tool) it is possible to tweak atom O3' of U63 so it sits better in density, moving the other two phosphate oxygen atoms:
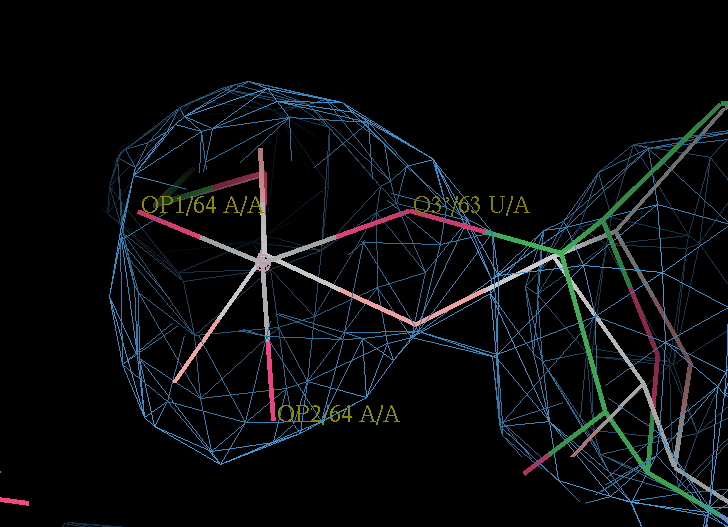
- doing this changes the epsilon torsion to a much better -93 degrees and pushes the sugar to a sensible looking pucker. From coot save the new model. This is my attempt.
- refine the revised model by:
refine -p 02_better_v3-coot-0.pdb -m 3g4m.mtz \
-d 03_after_U49_tweak -l NCO_correct.cif -l 2BP.grade_PDB_ligand.cif \
-M TLSbasic AnalyseVoidsLast=no \
TntDictionary_nuclgeo=`pwd`/nuclgeo_new_wiki_2011.dat > 03_after_U49_tweak.log
- compare results to pdb and 02 the straight refine.
- So tweaking U49 means that MolProbity geometry statistics are improved. After this the geometry scores are as good as or better than the pdb refinement. The Rfree goes up a little bit but Rfree can be noisy.
Using utility torsion restraints to favor canonical RNA puckers
- It is possible to further improve the geometry of the RNA by using utility torsion restraints to favor either a C3'-endo pucker or a C2'-endo pucker for each of the ribose rings in the RNA structure.
chmod a+x c3endo_or_c2endo_utiltor_restraints_all.pl
./c3endo_or_c2endo_utiltor_restraints_all.pl < 03_after_U49_tweak/refine.pdb > pucker_restraints.Gelly
- For each residue the jiffy chooses between restraints for a C3'-endo conformation or alternatively to a C2'-endo pcuker. It does this by evaluating the C1'-C2'-C3'-C4' torsion angle. Positive values are likely to indicate C3'-endo and negative C2'-endo.
- in this case 10 residues are restrained to C2'-endo:
grep "restraints to C2'-endo" pucker_restraints.Gelly
# resnam="U" chsnam="A" resnum="22" -ve torsion, restraints to C2'-endo
# resnam="U" chsnam="A" resnum="34" -ve torsion, restraints to C2'-endo
# resnam="A" chsnam="A" resnum="35" -ve torsion, restraints to C2'-endo
# resnam="U" chsnam="A" resnum="36" -ve torsion, restraints to C2'-endo
# resnam="U" chsnam="A" resnum="47" -ve torsion, restraints to C2'-endo
# resnam="U" chsnam="A" resnum="48" -ve torsion, restraints to C2'-endo
# resnam="U" chsnam="A" resnum="49" -ve torsion, restraints to C2'-endo
# resnam="G" chsnam="A" resnum="62" -ve torsion, restraints to C2'-endo
# resnam="U" chsnam="A" resnum="63" -ve torsion, restraints to C2'-endo
# resnam="A" chsnam="A" resnum="65" -ve torsion, restraints to C2'-endo
- the other 57 residues are restrained to C3'-endo
- The restraints file can be used with the autoBUSTER refine command line option -Gelly pucker_restraints.Gelly:
refine -p 03_after_U49_tweak/refine.pdb \
-m 3g4m.mtz -d 04_pucker_restraints \
-l NCO_correct.cif -l 2BP.grade_PDB_ligand.cif \
-M TLSbasic AnalyseVoidsLast=no \
TntDictionary_nuclgeo=`pwd`/nuclgeo_new_wiki_2011.dat \
-Gelly pucker_restraints.Gelly > 04_pucker_restraints.log &
- The Rfree is a little bit increased. But the geometry scores are improved.
- For 04 results, the prosit pucker parameter server http://cactus.nci.nih.gov/prosit/ classifies all residues as either C2'-endo or C3'-endo
- coot's "pukka pucker" only exception is for residue 64 that is held at a C3'-pucker but has an "inconsistent phosphate distance". Considering that bot the preceding and following residues are C2'-endo the next thing to try would be altering the restraints for this residue. This is left as an exercise for the reader!
Page by Oliver Smart original version 19 October 2011, revised 21 October. Address problems, corrections and clarifications to buster-develop@globalphasing.com