rhofit introductory tutorial on 2qtu
version: October 21 2010
Content:
- introduction
- starting files: autobuster refine -L run
- using grade to prepare a cif restraint dictionary for the ligand
- rhofit simple run
- getting rhofit to look for two binding sites with -xclusters 2
- getting rhofit to search through ligand chiral centres with -scanchirals
- starting an autobuster refinement with combined protein and rhofit best ligand positions
1. introduction
- This tutorial shows how rhofit can be used to fit a ligand to autoBUSTER difference density. grade is used to prepare the ligand dictionary.
- As an example we will use pdb entry 2qtu : this is the estrogen receptor beta ligand-binding domain binding a benzopyran ligand:

- This version of the tutorial was first written for the October 2010 release of rhofit and grade.
2. starting files: autoBUSTER refine -L run and ligand cif dictionaries
- All files to run the tutorial are contained in this tarball rhofit_2qtu_tutorial.tgz. Download this and unpack with command:
tar xzf rhofit_2qtu_tutorial.tgz
cd rhofit_2qtu_tutorial/
- rhofit requires a difference map with ligand density and a protein structure. It is designed to work well with autoBUSTER refine -L or refine -Lpdb options.
- For 2qtu the autoBUSTER refine -L run has already been done starting from a molecular replacement model supplied_molrep_2iog_2qtu.pdb that used as a starting model the related pdb entry 2i0g . The job was run with the options:
cat supplied_01_autobuster.source
# rhofit tutorial example 2qtu
# initial model is molecular replacement using 2i0g as search model
# refine this with -L to get ligand sites
( time refine \
-p supplied_molrep_2i0g_2qtu.pdb \
-m supplied_2qtu.mtz \
-RB -L -autoncs \
-d supplied_01_autobuster_d ) > & \
supplied_01_autobuster.log
- To save space the directory supplied_01_autobuster_d has been edited to just contain the final pdb and mtz files, required as input to rhofit.
3. using grade to prepare a cif restraint dictionary for the ligand}
- rhofit also needs to know what ligand to fit, in particular what a restraints dictionary that describe its conformational properties.
- grade is a method of producing refinement dictionaries that uses the mogul program from the CCDC together with semi empirical quantum chemical geometry optimization.
- we will use as an input the SMILES string from ligand expo entry for 3AS
- grade can be run for this:
grade 'COCc1cc(O)cc2[C@@H]3CC(F)(F)C[C@@H]3[C@@H](Oc12)c4ccc(O)cc4' \
-ocif 3AS_smi_grade.cif -opdb 3AS_smi_grade.pdb
- Note that the SMILES string has to be put in quotes to prevent it being mangled by the shell interpreter.
- The run takes about 80 seconds on my workstation.
- The refmac-style cif restraint dictionary produced will have the correct chemistry but it will not have the same atom names as the pdb ligand. If the correct atom names are wanted then used the grade_PDB_ligand tool should be used, with the pdb ligand code:
grade_PDB_ligand 3AS
- To show how rhofit can cope when the chirality of ligand atoms is not know we will use grade to produce a dictionary for 3AS with two of the chiral centres inverted (replacing @@ with @ in the first and last occurrence):
grade 'COCc1cc(O)cc2[C@H]3CC(F)(F)C[C@@H]3[C@H](Oc12)c4ccc(O)cc4' \
-ocif 3AS_invert2_grade.cif -opdb 3AS_invert2_grade.pdb
4. rhofit simple run
- here rhofit is run to find best single potential binding site and conformation for the ligand in the protein:
rhofit -l 3AS_smi_grade.cif \
-p supplied_01_autobuster_d/refine.pdb \
-m supplied_01_autobuster_d/refine.mtz \
-d tutorial4
- -l is used to specify the restraint dictionary that describes the ligand to be fitted. This can be in either refmac-compatible restraint dictionary or TNT restraint dictionary format (see rhofit manual "Describing the ligand" section)
- -p and -m are used to specify the protein structure and mtz file, as produced by autoBUSTER in section 2.
- -d is used to specify the output directory for results (see rhofit manual).
- This should run in about 2 minutes on a modern workstation.
- Results are written to the tutorial4 directory. So change directory by
cd tutorial4
- The visualise-rhofit-coot (see also $BDG_home/docs/rhofit/manual/index.html#visualise-rhofit-coot) utility can be used to conveniently view rhofit solutions with the coot program:
visualise-rhofit-coot
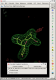
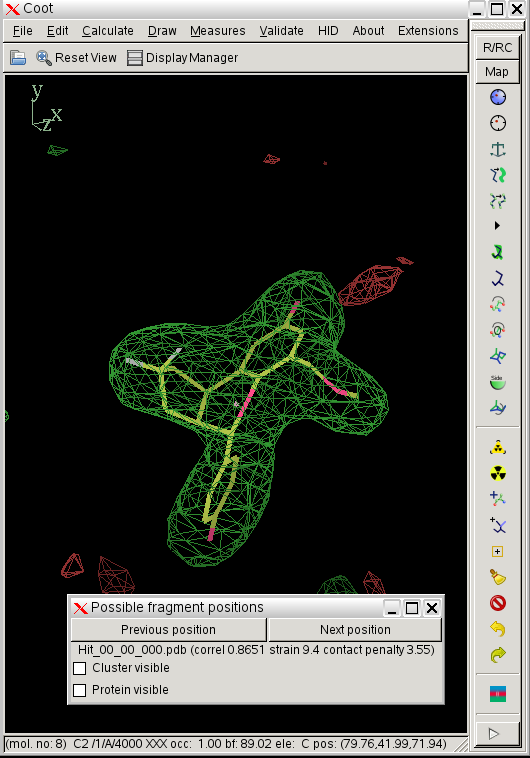
- in this case rhofit identifies two possible ligand binding modes in the A site:
 click on image to enlarge
click on image to enlarge- the two binding modes are close to one another and differ mainly in the pucker of the saturated ring with the two fluorine atoms bound. They are sufficently close that subsequent refinement is likely to lead to the same position.
- the most important results of the runs are summarized in the results.txt file in the results directory:
cat results.txt
rhofit version 1.2.0
Run in directory /home/osmart/2010/10/rhofit_2qtu_tutorial/rhofit_2qtu_tutorial with command-line
-l 3AS_smi_grade.cif -p supplied_01_autobuster_d/refine.pdb -m supplied_01_autobuster_d/refine.mtz -d tutorial4
Volume of cluster used for fitting: 148.6
rhofit ligand LigProt Poorly
total Correl strain contact fitting LigProt contact to residues
File Chain score coeff score score atoms (% means zero weighted in score)
=================================================================================================
Hit_00_00_000.pdb A -1672.1 0.8651 9.4 3.5 0/26 A|302:ALA A|377:PHE A|475:HIS A|476:LEU A|487:VAL
Hit_00_00_001.pdb A -1662.0 0.8623 12.8 11.0 0/26 A|302:ALA A|377:PHE A|476:LEU A|487:VAL
- See "What is in results.txt" section of rhofit documentation (also at $BDG_home/docs/rhofit/manual/index.html#results.txt).
- after doing this change directory back to the main tutorial directory.
5. getting rhofit to look for two binding sites with -xclusters 2
- As 2qtu has 2 fold ncs, there are two ligand binding sites. Un this example we will get rhofit to predict the ligand binding position in the two best "clusters" found.
- make sure that you are in the tutorial directory and enter the command:
(time rhofit -l 3AS_smi_grade.cif \
-xclusters 2 \
-p supplied_01_autobuster_d/refine.pdb \
-m supplied_01_autobuster_d/refine.mtz \
-d tutorial5
- this will run rhofit and place results in the directory tutorial5, cd to this directory.
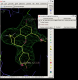
- The visualise-rhofit-coot utility can be used to quickly scroll through the solutions found. Try clicking on the "Protein visible" tick box to see the protein. Also switch between the different clusters using the "Previous search region" and "Next search region".
 click on image to enlarge
click on image to enlarge - In this case rhofit has found both the A site and B site binding positions for the ligand. It can be noted that the ligand in the A site (Hit_00_00_000.pdb) is supplied with the chain id A and that in the B site with chain id B (Hit_01_00_000.pdb). The B site contains two water positions that have not removed by autoBUSTER clustering, but rhofit disregards waters in assessing ligand protein contacts so is not badly affected (Click on the "Protein visible" button).
- Note, to help with further refinement rhofit outputs a file merged.pdb that combines the protein with what it thinks is the best position for each site, see section 7.
6. getting rhofit to search through ligand chiral centres with -scanchirals
- As well as being able to search for multiple binding sites rhofit is able to cope with the situation where you are not exactly sure that your ligand model has correct chirality, provided that you have density good enough to distinguish between the possibilities. For the purposes of this tutorial a refmac-compatible cif restraint dictionary for the 3AS ligand with two inverted chiral centres (out of three) was produced (see section 3).
- rhofit can be used to systematically search through the possible chiral combinations in the best cluster identified:
rhofit -l 3AS_invert2_grade.cif \
-scanchirals \
-xclusters 2 \
-p supplied_01_autobuster_d/refine.pdb \
-m supplied_01_autobuster_d/refine.mtz \
-d tutorial6
- This takes abourt 40 minutes on my workstation.
- The results directory tutorial6. contains lots of files (see "What is in results.txt") . Importantly.
- visualise-rhofit-coot can be used to quickly scroll through the solutions found. Following a -scanchirals run the different chiral combinations can be scrolled through using the"Previous search region" and "Next search region" buttons. The "Previous position" and "Next position" buttons switch between different fits for a particular chiral combination.
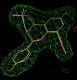
- In this case the best solution has a correlation coefficient of 0.87 and has the same chiral centres as the ligand from 2qtu.pdb (shown in purple):
 click on image to enlarge
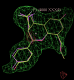
click on image to enlarge- Similarly for the B site the same chiral combination is found:
 click on image to enlarge
click on image to enlarge- rhofit thinks the next best chiral combination for the A site has a correlation coefficient of 0.83 (marginally lower) and atom C12 inverted:
 click on image to enlarge
click on image to enlarge
- As well as the -scanchirals rhofit has a -nochirals option. This is much faster in that it turns off all the chiral restraints. Chiral atoms will still remain chiral because of the bond angle restraints around the atom but can invert at will during the fit. The -nochirals option works in this case but is less systematic and could be fooled.
- you clearly need good density to be able to determine chiralities from difference density!
7. starting an autobuster refinement with combined protein and rhofit best ligand positions
In this case there are two ligand binding sites in the protein. In part 5 a merged pdb file is created by rhofit that has the protein with two best ligand binding positions and water molecules that do not clash with the ligands.
- The merged pdb can be refined autoBUSTER:
( time refine \
-p tutorial5/merged.pdb \
-m supplied_2qtu.mtz \
-autoncs -WAT \
-l tutorial5/best.cif \
-d tutorial7 ) > \
tutorial7.log
- note the use of the rhofit output TNT dictionary for the ligand best.dic. This is particularly important if you have searched for chiralities.
- -autoncs restraints will include the ligand as rhofit calls the ligand in the A site residue A 4000 and that in the B site B 4000.
- -WAT water insertion is used as we have removed the waters from the protein.
- Use coot to compare the final protein with fitted ligand with the 2qtu.pdb entry. There are only minor differences mainly in the positioning of the ether group that is not well determined at this resolution.
Page by Oliver Smart and Andrew Sharff, October 2010. Address problems, corrections and clarifications to buster-develop@globalphasing.com
 click on image to enlarge
click on image to enlarge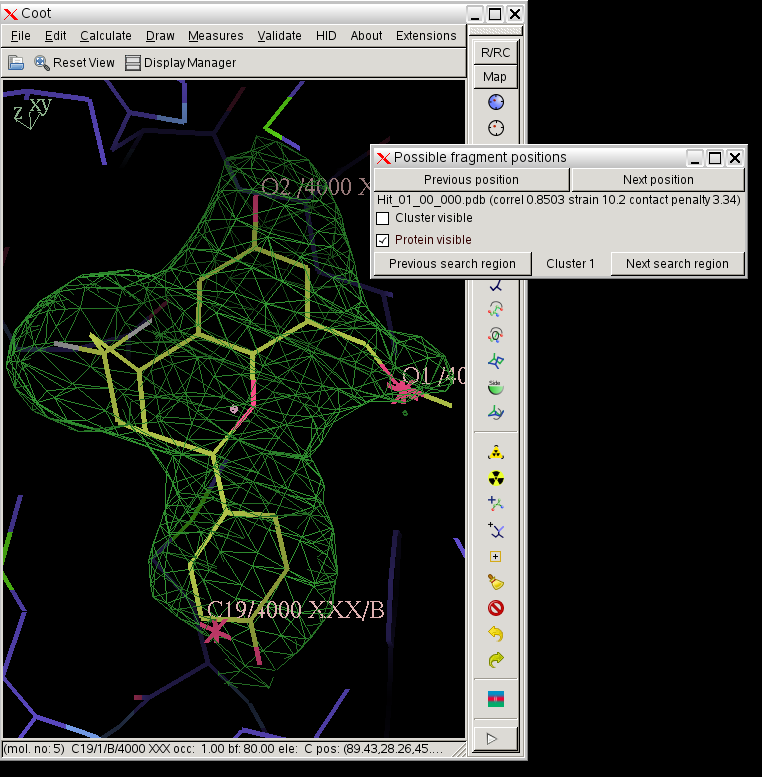 click on image to enlarge
click on image to enlarge 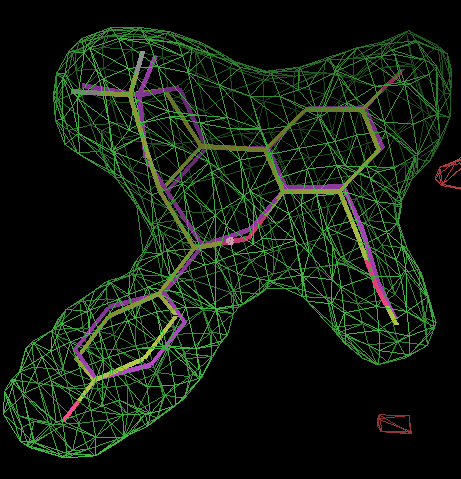 click on image to enlarge
click on image to enlarge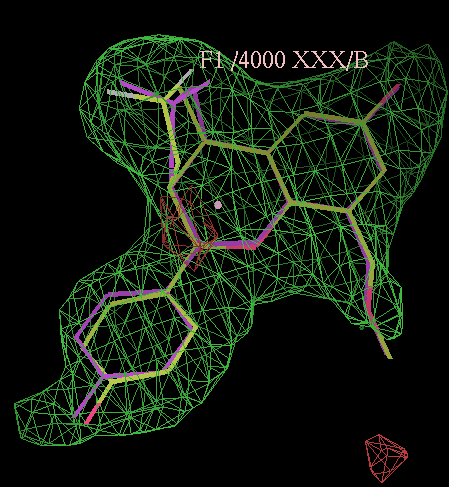 click on image to enlarge
click on image to enlarge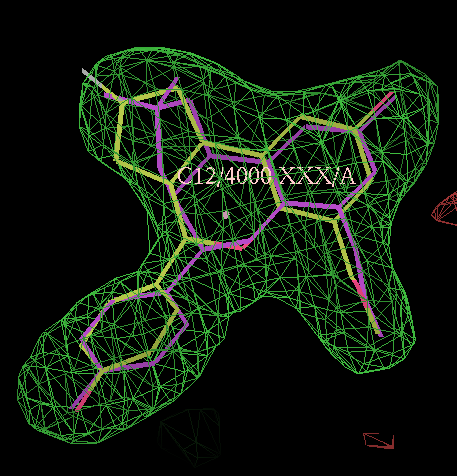 click on image to enlarge
click on image to enlarge