BUSTER -forcefield option
contents
- Introduction
- How to set up OpenEye buster_helper_mmff
- The RDKit helper
- Using the -forcefield option
- Specification of which ligand to represent with MMFF
- Checking the force field helper has correctly interpreted the ligand chemistry
- Altering the weight used for -forcefield
- Using the -forcefield option
- Known limitations and issues
- Tutorial
Introduction
The -forcefield refine option allows the use of a force field to represent a ligand. It uses the procedures developed for the Direct use of weighted Quantum Chemical Energy for ligands in BUSTER refinement. But instead of using a quantum chemical representation for the ligand a molecular mechanics force field is used. Initial work has concentrated on providing helpers for the MMFF94 force field (that is widely used and well regarded). Force fields offer a number of advantages compared to quantum methods:
- They are robust. Quantum methods have a tendency to fail for distorted or highly charged systems.
- They are computationally cheap.
- A well parameterized force field (like MMFF94) will give accurate results for molecules similar to those in its "training set". In these cases strain energy results comparable to computationally expensive accurate DFT calculations can be obtained (to be published).
- Force fields are extensively used in computational chemistry so results obtained from X-ray crystallography will be "computation ready". Furthermore, a high resolution X-ray complex including the ligand should validate the force field used (if a ligand is indicated to be highly strained then this would often indicate that the force field needs improvement.
The OpenEye implementation of MMFF94 includes extensions enabling the forcefield to represent compounds containing selenium (details) or boron (details). MMFF94 is used in the OpenEye AFITT automated ligand fitter.
How to set up OpenEye buster_helper_mmff
- To use the OpenEye buster_helper_mmff you must have an AFITT installation that includes the executable and a working license file. You will also need a recent BUSTER release, dated 26 Nov 2013 or later
- The environment variable BDG_TOOL_OE_HELPER_BIN_DIR must be set to the directory that contains the buster_helper_mmff executable.
- The recommended way to do this is edit (or create) the files:
$BDG_home/setup_local.sh $BDG_home/setup_local.csh
- where $BDG_home is the root directory of the Global Phasing Software setup.
- Please see the detailed installation instructions that can be found in $BDG_home/docs/installation/index.html.
- Once this is done the -forcefield option will automatically invoke the buster_helper_mmff helper.
The RDKit helper
- For users who do not have AFITT we are working a helper that uses the RDKit http://rdkit.org/ implementation of MMFF94 written by Dr Paolo Tosco http://www.personalweb.unito.it/paolo.tosco/
- There are some issues with charged ligands that need to be solved before it can be released.
Using the -forcefield option
Specification of the ligands to represent with MMFF
- The forcefield option can be used with refine and gelly_refine if there is a helper available. If your ligand has the three letter residue code LIG, to use an MMFF94 forcefield for it in BUSTER refinement:
refine -p model.pdb -m data.mtz -d 01 -l grade-LIG.cif -forcefield LIG > 01.log
- If there is more than one instance of LIG in the structure (for instance because of NCS), a separate "force field group" will be created for each one.
- If you want to use the MMFF94 for more that one type of ligand then you can do this by providing a comma-separated list, for instance -forcefield LIG,INH
- Please note that a cif restraint dictionary is still required for the ligand. This dictionary is absolutely essential because the bond connectivity and bond orders are crucial for the helper to be able to correctly set up the MMFF94 force field.
- The ligand must have had hydrogen atoms added if appropriate. Hydrogen atoms are necessary both for the MMFF94 force field to be valid and also for the helper to be able to correctly set up the molecular geometry. For this reason the -forcefield option will stop if presented with a ligand that lacks hydrogen atoms. If your ligand actually lacks hydrogen atoms use the -qm_allow_0_h option.
- It can be noted that the -forcefield option is used in the same way as the -qm option (see QM command line options wiki page) except the total ligand charge need not be specified. In fact the -forcefield option shares code with -qm and so the two options cannot be used at the same time.
Checking that the force field helper has correctly interpreted the ligand chemistry
- It is important to check that the forcefield helper has correctly identified the chemistry of the ligand. To do this the MMFF helpers (both OpenEye and RDKit) report the SMILES string of the ligand as part of the BUSTER refine output file LIST.html. For instance if you ran
refine -p model.pdb -m data.mtz -d rund -l grade-LIG.cif -forcefield LIG > run.log
- the first LIST.html file would be written to
run/01-BUSTER/Cycle-1/LIST.html
- For instance for the 2i0g tutorial example the file LIST.html would be obtained using the OpenEye helper. The part we are interested in looks like:
QM_HELPER_LOG: forcefield.pl invoking OpenEye Helper with /mnt/scratch_fs1/osmart/autobuster/Server/scripts/qm-mm-helpers/OpenEye.pl 0 1 AUTO
QM_HELPER_LOG:
QM_HELPER_LOG: gelly helper script to run OpenEye helpers for force field or QM energy/gradient calculation
QM_HELPER_LOG: Please obtain helpers for OpenEye http://www.eyesopen.com/
QM_HELPER_LOG: helper script location /mnt/scratch_fs1/osmart/autobuster/Server/scripts/qm-mm-helpers/OpenEye.pl
QM_HELPER_LOG: picked up charge=0 multip=1 method=AUTO from command line
QM_HELPER_LOG: Open Eye executable used: /home/osmart/2013/09/OpenEyeHelper/openeye/bin//buster_helper_mmff
QM_HELPER_LOG: :jGf:
QM_HELPER_LOG: :jGDDDDf:
QM_HELPER_LOG: ,fDDDGjLDDDf, BUSTER HELPER MMFF
QM_HELPER_LOG: ,fDDLt: :iLDDL;
QM_HELPER_LOG: ;fDLt: :tfDG;
QM_HELPER_LOG: ,jft: ,ijfffji, :iff
QM_HELPER_LOG: .jGDDDDDDDDDGt.
QM_HELPER_LOG: ;GDDGt:''':tDDDG,
QM_HELPER_LOG: .DDDG: :GDDG.
QM_HELPER_LOG: ;DDDj tDDDi
QM_HELPER_LOG: ,DDDf fDDD, Copyright (c) 2013
QM_HELPER_LOG: LDDDt. .fDDDj OpenEye Scientific Software, Inc.
QM_HELPER_LOG: .tDDDDfjtjfDDDGt
QM_HELPER_LOG: :ifGDDDDDGfi. Version: 2.3.0.4
QM_HELPER_LOG: .:::. Built: 20130710
QM_HELPER_LOG: ...................... OEChem version: 1.9.2 20130710
QM_HELPER_LOG: DDDDDDDDDDDDDDDDDDDDDD Platform: Ubuntu-12.04-g++4.6-x64
QM_HELPER_LOG: DDDDDDDDDDDDDDDDDDDDDD
QM_HELPER_LOG:
QM_HELPER_LOG:
QM_HELPER_LOG:
QM_HELPER_LOG: Input ISM: c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
QM energy for QMset 1 picked up as 316.47988
- Note that the helper reports the ligand SMILES string in the line(s)
QM_HELPER_LOG: Input ISM: c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
- in this case the SMILES string is
c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
- It is recommended that you check the chemistry by using your favorite SMILES to 2D diagram tool.
- For instance using Daylight Depict http://www.daylight.com/daycgi/depict
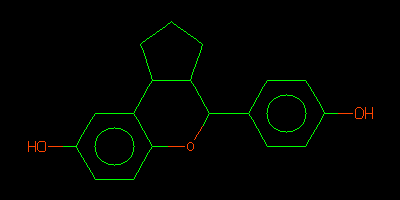
- Or using OpenBabel
egrep ISM rund/01-BUSTER/Cycle-1/LIST.html | head -1 | sed -e 's/.*ISM..//' > check.smi obabel check.smi -Ocheck.svg convert check.svg check.png
- to give
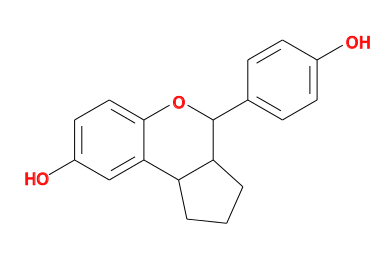
- The diagram can be compared to the expected ligand chemistry. In this case the I0G ligand is described at http://ligand-expo.rcsb.org/reports/I/I0G/index.html and the helper interpretation is correct.
- What to do if you get a SMILES string/diagram that is incorrect:
- check the bond orders as defined in the cif dictionary you are using for the ligand. A good way to do this is to use a recent version of coot as this displays them. The bond orders are found in the _chem_comp_bond.type field. It is worth trying Kekule type definitions (with explicit single and double bonds in place of aromatic.
- ask for help buster-develop@globalphasing.com we are actively trying to improve/support force field use.
Altering the weight used for -forcefield
- To alter the weight used for the MMFF force field use the --qm_weight Z command line option (See AutobusterLigandQMcommandLineOptions). The default weight is 16 and this generally works well.
Known limitations and issues
Currently the -forcefield option and helpers have the major limitation that ligands must be chemically complete with all hydrogen atoms. This limitation has consequences for handling both covalent and incomplete ligands.
Incomplete ligands
Ligands that are incomplete (have missing atoms) will not be handled properly at present. Any problem should be apparent by checking the SMILES string reported by the helper ISM line. It should be possible to fix this limitation in a future release.
Ligands that are covalently bound to the protein or other molecules
The current procedures will not properly handle covalently-bound ligands including proteins containing modified amino acids or nucleic acids with modified bases. Any problem should be apparent by checking the SMILES string reported by the helper ISM line. It should be possible to fix this limitation in a future release.
Ligands with alternate position atoms
The current procedure does not handle normal alternate positions in ligands. It is possible to treat alternates using a workaround, if you would like details contact buster-develop@globalphasing.com
It should be possible to fix this limitation in a future release.
grade S=O bond issue
Currently grade will incorrectly set the type for S=O bonds to "single". This will result in an incorrect force field setup, this will be shown by checking the SMILES string reported by the helper ISM line. For instance for the 1YU ligand from structure 4lxm the ISM SMILES produces a structure:
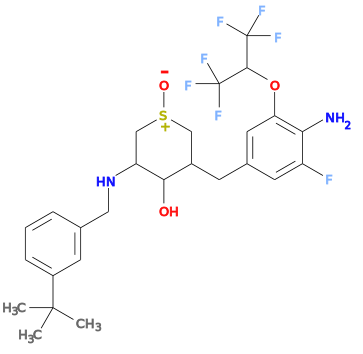
- Correcting the grade dictionary by setting S28 O32 bond to double rather than single produces a correct setup.
Tutorial
| Back to Ligand QM/force field top index page |
Page by Oliver Smart Nov 2013, modified June 2014 Any questions regarding our software or this wiki should be directed to buster-develop@globalphasing.com