WORK-IN-PROGRESS
Content:
Introduction
6Y7M: Zhang, L., Lin, D., Hilgenfeld, R. (2020). Crystal structure of the complex resulting from the reaction between the SARS-CoV main protease and tert-butyl (1-((S)–3-cyclohexyl-1-(((S)4(cyclopropylamino)–3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate. To be published.
Data
This is a mmCIF-only structure - so we need to do some small detour: 1
wget https://files.rcsb.org/download/6Y7M.cif sed -i "s% AAA % A %g" 6Y7M.cif gemmi convert 6Y7M.cif pdb6y7m.ent gzip pdb6y7m.ent fetch_PDB 6Y7M
The restraint dictionary for the ligand can be generated via
grade_PDB_ligand OEWor using our GRADE webserver.
Initial (re-)refinement
A series of "standard" refinement steps:
cd 6Y7M refine \ -p 6y7m.pdb \ -m 6y7m.mtz \ -RB \ -l OEW.grade_PDB_ligand.cif \ -d 01 | tee 01.lis refine \ -p 01/refine.pdb \ -m 6y7m.mtz \ -M TLSbasic \ -l OEW.grade_PDB_ligand.cif \ -d 02 | tee 02.lis refine \ -p 02/refine.pdb \ -m 6y7m.mtz \ -M TLSalternate -TLS \ -WAT \ -l OEW.grade_PDB_ligand.cif \ -d 03 | tee 03.lis
gives us
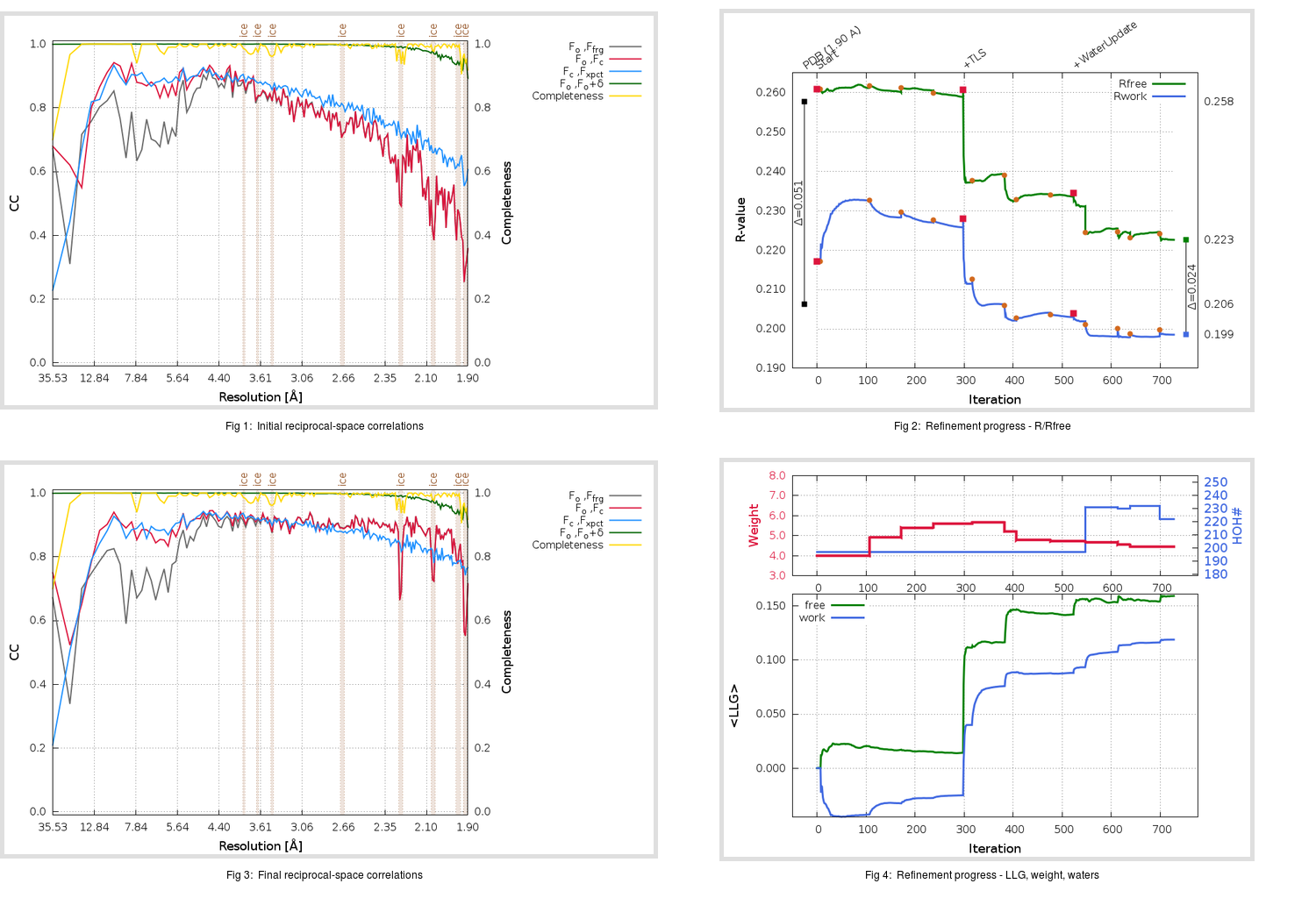 |
and going from
Ramachandran outliers = 0.33 %
favored = 96.71 %
Rotamer outliers = 3.41 %
C-beta deviations = 0
Clashscore = 3.78
RMS(bonds) = 0.0145
RMS(angles) = 1.94
MolProbity score = 1.78
to
Ramachandran outliers = 0.00 %
favored = 98.03 %
Rotamer outliers = 3.03 %
C-beta deviations = 1
Clashscore = 2.10
RMS(bonds) = 0.0113
RMS(angles) = 1.56
MolProbity score = 1.35
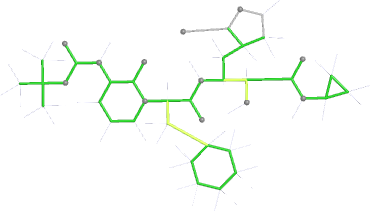
Looking explicitly at the ligand (using buster-report - see also here 2 ): below are some color-coded plots highlighting particular geometries that the CSD thinks (via Mogul) are suspect ('bad' = purple, 'poor' = violet, 'ok' = lime, 'good' = green, 'unknown' = gray).
| Stage | deposited (OCC=0.80) | initial (re-)refined (OCC=0.80) |
| bonds | 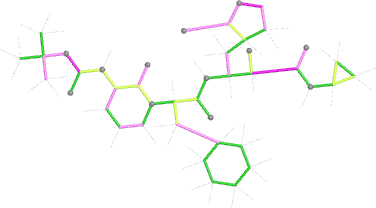 |
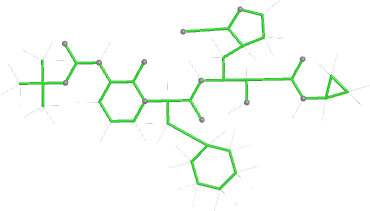 |
| angles | 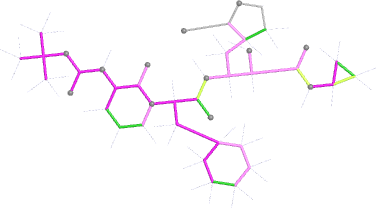 |
 |
| torsion | 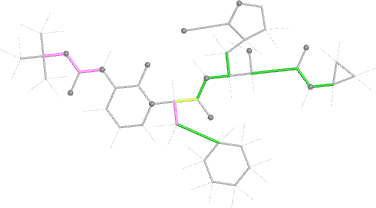 |
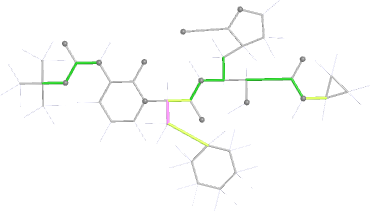 |
| density 3 | 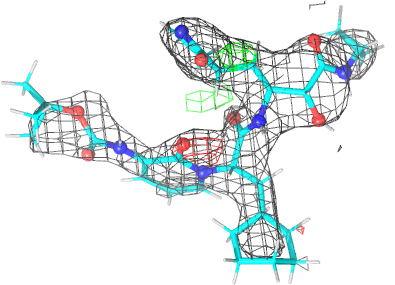 |
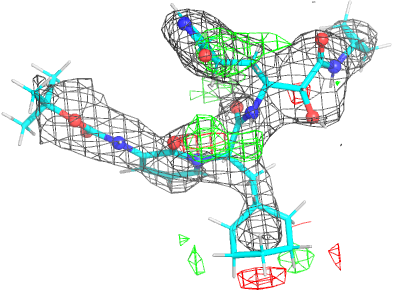 |
Full buster-report output and files
- deposited:
- (re-)refinement:
| Note | The remaining difference density after refinement is still being investigated. |
|---|
(Re-)fitting the ligand
To get a second opinion about the fit of the ligand to density, we can use rhofit after stripping th ligand out of the latest model
grep -v "OEW" aB_refine.01/03/refine.pdb > aB_refine.01/03/refine_noOEW.pdband using our ligand-detection feature in BUSTER:
refine \ -p aB_refine.01/03/refine_noOEW.pdb \ -m 6y7m.mtz \ -TLS \ -L \ -M MapOnly \ -d aB_refine.01/minusL.01 | tee aB_refine.01/minusL.01.lis
The resulting electron density map can be used to fit the compound in:
rhofit \ -l OEW.grade_PDB_ligand.cif \ -m aB_refine.01/minusL.01/refine.mtz \ -p aB_refine.01/minusL.01/refine.pdb \ -d rhofit.01 | tee rhofit.01.lis
The final PDB file (rhofit.01/merged.pdb) needs two small changes:
- adding the correct LINK card back in (could also be done in Coot) since this is a covalent ligand with a bond between SG of Cys-A145 and the C57 carbon of OEW
- setting the occupancy of all OEW atoms to 0.99 (as a marker for pdb2occ for occupancy refinement
We can now also create a correct restraints dictionary for the covalent linkage using aB_covalent_ligand:
aB_covalent_ligand rhofit.01/merged_edit.pdbwhich produces the required restraints and also shows how to use them in subsequent BUSTER refinements.
Combining all this into a new refinement command:
pdb2occ -p rhofit.01/merged_edit.pdb -o rhofit.01/merged_edit.occ refine \ -p rhofit.01/merged_edit.pdb \ -m 6y7m.mtz \ -TLS \ -WAT -M WaterUpdate2ndShell \ -l OEW.grade_PDB_ligand.cif \ -Gelly rhofit.01/merged_edit.occ \ MakeLINK_LinkagesFile=CYS-SG_OEW-C57.dat \ -l CYS-SG_OEW-C57.dic \ RunBusterDuplicatesOverride=CYO \ -d aB_refine.01/04 | tee aB_refine.01/04.lis
And looking again at the ligand geometry (in comparison with the initial refinement):
| Stage | initial (re-)refined (see above) | (re-)fitted and refined (OCC=0.66) |
| bonds | |
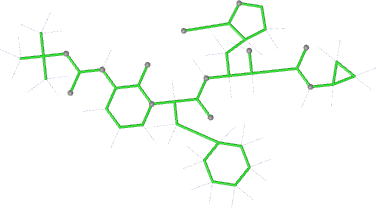 |
| angles | |
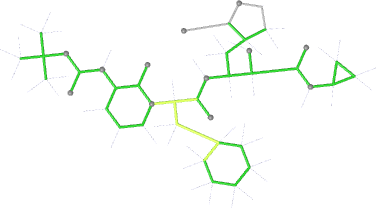 |
| torsion | |
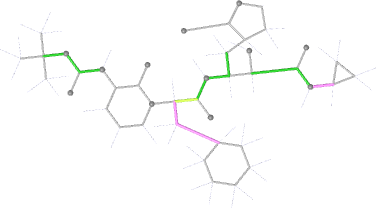 |
| density | |
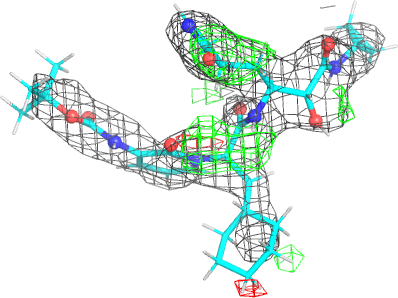 |
Files
Footnotes
- *1: we will need to simplify this with better mmCIF support in future BUSTER releases
- *2: since 11/2019 this is also part of the wwPDB validation
- *3: the orientation of the view is defined automatically by buster-report and can be different for different refinements - due to small conformation changes