rhofit documentation |
|
Copyright © 2009-2017 Global Phasing Limited
All rights reserved.
This software is proprietary to and embodies the
Authors: (2009-2011) O.S. Smart, T.Womack |
Rhofit is a tool for fitting ligands into difference density. It differs from FitMAP (our previous fitting tool) in that it is able to change bond lengths and angles within the ligand, rather than only rotate torsion angles; this also allows it to search for correct ring conformations for macrocycles. In addition, rhofit can also search for the correct chirality if this is not known for an input ligand.
Rhofit uses the gelly geometry function to assess ligand strain and protein-ligand contacts, so its results will be compatible with further BUSTER refinement. Rhofit runs a large number of independent trials and reports the best solutions that it finds; you can give it some idea of how long you want it to search for. We aim for rhofit with default parameters to take significantly less time that the refinement required to produce its input.
Take some structure in which there is a ligand to be found and do
refine -L -p structure.pdb -m data.mtz -d 00 | tee 00.listo tell BUSTER to try ligand-chasing.
There are a fair number of command-line arguments available, but the easiest use cases are
By default, rhofit will produce a list of the connected blobs of difference density from the input MTZ file which are large enough to contain the ligands (each blob is described in a cluster file). rhofit will then try quick runs to fit the ligand into each of these blobs, and then try much more intensively to fit the ligand into the single blob into which it seemed to fit best. If you expect there to be more than one copy of the ligand bound to the structure, use the -xclusters option.
After the rhofit command has finished, it will create a directory 'rhofit'. The easiest way to explore this directory is with the tool visualise-rhofit-coot, which lets you look through the list of positions that rhofit has found for your ligand; they are in order of correlation coefficient so generally the correct conformation should be the first one that appears. The section below interpreting rhofit output has more information.
Please see rhofit introductory tutorial on 2qtu Wiki Page
Planes, particularly large planes, are extremely strong restraints; they make motion away from the plane almost impossible even when the density extremely strongly requests it, and the worst examples of bad fits that we see are due to bad planes in the input dictionary. rhofit-1.2 attacks this problem from two directions:
At the end of the fitting process, planes which have been turned off are deleted from the best.cif file produced by rhofit — so a refinement of merged.pdb using best.cif as dictionary will not reinstate the plane.
There is a command-line option -trustplanes which turns off the plane-breaking and torsion-plateauing machinery; we would suggest that this is used only in cases where the machinery gives wrong answers, and buster-develop@globalphasing.com would be very interested to hear as much as you are allowed to reveal about cases which fail like this.
rhofit looks for a line of the form
# BUSTER-KEYWORD keyword1 keyword2 ...
in the input
dictionary, and changes its behaviour when it sees certain keywords:
| TRUSTCOORD | The coordinates in the chem_comp_atom.{x,y,z} fields in the CIF file are taken to be the result of a careful generation from the restraints, and rhofit uses them directly rather than regenerating from the restraints |
| TRUSTTORS | The restraints in the chem_comp_tor section of the dictionary are considered to be of good quality, and are used when doing the fitting |
At present grade is the only tool which writes out these keywords.
| -h | Quick help message listing most important arguments. |
| -l ligand.dic | Restraint dictionary for ligand. This can be either in CCP4-style CIF format or TNT format. A good restraint dictionary is important to get good fits. We suggest that the grade tool is used to prepare dictionaries for rhofit. |
| -keepH | This option indicates that rhofit should keep hydrogen atoms in the fit. To use this you must specify an input restraint dictionary with hydrogen atoms in it. By default rhofit will strip hydrogen atoms. Using hydrogen atoms will slow down rhofit and may worsen sampling of things like saturated rings. |
| -nochirals | rhofit will ignore all CHIRAL cards in the input dictionary. This option allows chiral centres to invert at will during the run. Note that atoms will remain tetrahedral because of the bond angle restraints around the centre. This option is a "quick and dirty" one - use -scanchirals or -scanchiralsboth, if you have time |
| -scanchirals | rhofit will try fitting with every possible permutation of chiralities, and work out which produces the best fit. Note that this will try large numbers of permutations if the ligand has many chiral centres: 256 permutations for a ligand containing two ribose rings. For these cases -nochirals, -scanchiralsboth or -scanchiral <c> should be tried first. |
| -scanchiralsboth | This is similar to -scanchirals, but only chiralities marked as "both" in dictionary will be sampled. |
| -scanchiral <c> | This is similar to -scanchiralsboth, but giving an explicit (comma-separate) list of chiral center atom names. |
| -lp ligand.pdb | This option can be used to seed the rhofit process with a particular starting position. |
| -trustplanes | By default, if the density disagrees strongly with a plane in the input dictionary, rhofit will turn it off - see the plane-handling section above. |
| -m refine.mtz | MTZ file to use for difference density. This can be produced by either BUSTER or REFMAC. (the default is refine.mtz in the current directory) |
| -p refine.pdb | PDB file to use for determining protein/ligand clashes (the default is refine.pdb in the current directory) |
| -clusterweight x | Set weight used for difference density score in rhofit to x (default 0.25). This does not normally need adjustment. |
| -clashweight x | Set weight used for ligand-protein clash score in rhofit to x (default 0.5). To turn off ligand-protein contacts use -clashweight 0.0 This parameter seldom needs adjustment. |
| -sctrimb x | This adjusts the B range threshold used to assess whether a protein side chain is poorly placed in density and so should be disregarded when assessing ligand-protein clashes. By default a value of 30 Angstroms squared is used. To turn the feature off set to a large value like -sctrimb 1000.0 |
|
rhofit uses an intermediate file called a "cluster" file to define the region
to fit. This is a pseudo pdb file and can be displayed using the visualise-rhofit-coot visualiser.
By default rhofit will itself work out potential ligand binding sites and their cluster files.
It will then quickly assess in which single cluster the ligand fits best and
thoroughly find out the exact ligand fits in this single site. This behaviour can be controlled by
the following options: |
|
| -xclusters n | Use the best n clusters, found by rhofit, in which the ligand seems to fit best. So if you have a trimer each of which has a ligand binding site specify -xclusters 3 |
| -allclusters | Find ligand binding fits for every cluster found by rhofit. |
| -c cluster.pdb | Filename of a single cluster in which to fit the ligand. This option stops rhofit from finding clusters itself. cluster files are produced by BUSTER ("refine"), rhofit itself and the prep_rhofit utility (get help for this with prep_rhofit -h). Only use this option if rhofit itself does not correctly identify the binding site you are interested in. |
| -C cluster.lis | Filename of a file containing a list of clusters in which to try to fit the ligand. |
| -C cluster.lis | Filename of a file containing a list of clusters in which to try to fit the ligand. |
| -prf.c cut1,cut2 -prf.V vol | You can use these options to directly control the prep_rhofit run made by rhofit to find clusters from the density. Note that you must specify both -prf.c and -prf.V they will not work separately. -prf.c cut1,cut2 is used to specify the values for prep_rhofit -c: this is the "significance cut-off values [rms] for Fo-Fc map (cut1) and 2Fo-Fc map (cut2)". Default values are cut1=3.0 and cut2=1.0. Note that a comma must be used to separate the two values for -prf.c. -prf.V vol is used to control the volume used in prep_rhofit -v. There is no default volume in rhofit. Instead rhofit estimates the volume from the number of atoms in the ligand and then adjust this if no clusters are found. |
| -thorough | Run more trials than usual in the rhofit fit. This may be useful if you want to exact fits for large complex ligands. This is an option to use in pipelines where you have large amounts of compute power available and are not waiting impatiently for the result |
| -quick | Run three times fewer trials than usual; note that this may not find good conformations for complicated ligands. But if you want a quick result and are happy to tweak the result in coot use this option (and also consider -nocorrel and -nochiral). |
| -nocorrel | By default after fitting each solution is assessed for its correlation coefficient against Fo-Fc. The -nocorrel option saves times by not doing this. |
| -correlsort | The first release of rhofit reordered hits in order of corrrelation coefficient against Fo-Fc rather than using the rhofit total score which takes in account ligand strain and ligand-protein contacts. The -correlsort will reorder hits on the basis of CC alone. This is usually not a good idea as ligand strain is then ignored. |
| -randskip int | The rhofit trial process uses random numbers to produce randomized conformations/fits. By default the same random number series is used on a rerun of a job (to allow adjustment of conditions such as weights to not be made more difficult). If a different set of random numbers is wanted specify -randskip integer_number where integer_number is an integer value e.g., 57 or 2341. This should produce slightly different results. |
| -d directory | Directory to put all final results of the run (defaults to ./rhofit). |
| -td directory | Directory for temporary data during rhofit run. By default a directory will be created (in $TMPDIR if that variable is set, or /tmp otherwise) and removed at the end of a run. -td is only likely to be useful in diagnosing problems |
| -resnum N | Specifies the residue number to be used for the output hit and pdb files. A default value of 4000 is used. N must be an integer value in the range -999 to 9999. Note that if two sites are found close to a single chain then the residue number is incremented by one for the second site. |
| -hitmaxn int | The maximum number of hits to be output by rhofit. If you are only interested in the top hit set the value to one. The default value is 10. Must be set to a positive integer value. |
| -hitmaxd d | Sets the distance d used to assess whether two hits are the same. The maximum displacement distance for any atoms between the positions is found. If this is less than d then the positions are regarded as identical and only the with best rhofit total score is output (this is a greedy clustering). By default d is set to 1.5 Å. If you think that rhofit outputs hits that are two similar then try increasing the value to 3.0. |
| -hitfncut f | Sets the function value cut off beyond which rhofit thinks that you will not be interested in seeing hits. If a hit has a rhofit total score that it worse than f units compared to best hit it is not output. A default value of 30.0 is used. To see hits thats rhofit regards as poor increase the value of f and -hitmaxn. |
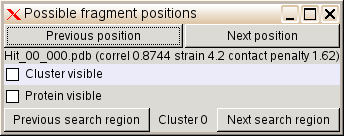
rhofit produces an output directory whose name you can specify with the -d option at the end of the run this will contain the result. The ligand fit solutions are best examined using the visualise-rhofit-coot utility. It allows you to use the coot program to quickly click thorough potential fits of the ligand to difference density. To start the utility first cd into the results directory and type the command visualise-rhofit-coot (you must have both coot and the Global Phasing setup.csh or setup.sh defined). The command will start coot and present you with the best solution found by rhofit and a dialogue box like:
To examine the different hits for this cluster then use the Previous position and Next Position buttons. The Previous search region and Next search region buttons allow switching between different clusters or chiral combinations. The check boxes Cluster visible and Protein visible allow you to quickly turn on/off cluster and protein display
The results.txt provides a summary of the rhofit run . A typical file (example 6 from the rhofit wiki 2qtu tutorial) is:
Summary of results (also written to rhofit_tutorial4/results.txt)
rhofit version 1.2.0
Run in directory /mnt/scratch_fs1/twomack/rhofit_2qtu_tutorial with command-line
-l supplied_ligand_wrongchiral_libcheck_XXX.cif -xclusters 2 -p supplied_01_buster_d/refine.pdb -m supplied_01_buster_d/refine.mtz -d rhofit_tutorial4 -scanchirals
Volume of clusters used for fitting: 148.6 138.6
rhofit ligand LigProt Poorly
total Correl strain contact fitting LigProt contact to residues
File Chain score coeff score score atoms (% means zero weighted in score)
=================================================================================================
Hit_00_00_000.pdb A -1693.0 0.8724 -5.3 3.7 0/26 A|476:LEU A|487:VAL
Hit_00_01_000.pdb A -1646.2 0.8512 12.8 5.1 0/26 A|356:PHE A|376:ILE A|476:LEU A|487:VAL
Hit_00_02_000.pdb A -1604.6 0.8465 44.0 9.7 0/26 A|376:ILE A|476:LEU A|487:VAL
Hit_00_02_001.pdb A -1603.2 0.8330 20.4 13.1 0/26 A|299:THR A|302:ALA A|376:ILE A|377:PHE A|475:HIS A|476:LEU A|487:VAL
Hit_00_02_002.pdb A -1595.2 0.8088 13.5 17.7 0/26 A|299:THR A|302:ALA A|475:HIS A|476:LEU A|487:VAL
...
Hit_01_00_000.pdb B -1469.6 0.8533 8.8 5.1 0/26 B|299:THR B|302:ALA B|475:HIS B|476:LEU B|487:VAL W|26:HOH% W|6:HOH%
Hit_01_00_001.pdb B -1446.3 0.8501 11.4 16.4 0/26 B|298:LEU B|299:THR B|302:ALA B|475:HIS B|476:LEU W|26:HOH% W|6:HOH%
Hit_01_01_000.pdb B -1427.8 0.8314 27.9 4.8 0/26 B|302:ALA B|305:GLU B|476:LEU B|487:VAL W|26:HOH% W|6:HOH%
Hit_01_01_001.pdb B -1420.5 0.8362 31.0 9.4 0/26 B|299:THR B|302:ALA B|305:GLU B|475:HIS B|476:LEU B|487:VAL W|26:HOH% W|6:HOH%
Hit_01_01_002.pdb B -1413.3 0.8324 19.1 12.6 1/26 B|298:LEU B|299:THR B|305:GLU B|475:HIS B|476:LEU W|26:HOH% W|6:HOH%
Hit_01_01_003.pdb B -1413.2 0.8412 24.2 22.5 0/26 B|298:LEU B|299:THR B|302:ALA B|305:GLU B|475:HIS B|476:LEU W|26:HOH% W|6:HOH%
Hit_01_02_000.pdb B -1402.5 0.8312 24.1 21.7 0/26 B|298:LEU B|299:THR B|302:ALA B|305:GLU B|376:ILE B|475:HIS B|476:LEU W|26:HOH% W|6:HOH%
Hit_01_02_001.pdb B -1402.5 0.8216 40.2 9.5 0/26 B|302:ALA B|305:GLU B|376:ILE B|476:LEU W|26:HOH% W|6:HOH%
Hit_01_02_002.pdb B -1400.7 0.8358 33.5 17.2 0/26 B|298:LEU B|299:THR B|305:GLU B|376:ILE B|475:HIS B|476:LEU W|26:HOH% W|6:HOH%
...
|
| results.txt | A text file that provides a summary of the run (see above for discussion). |
| best.pdb | The best position found for the ligand in the best cluster. |
| merged.pdb | Combination of the protein with the best hit for the ligand in each cluster found. Water molecules that clash with the ligand are stripped out, others left alone |
| refine.pdb | The protein used in the rhofit run |
| refine.mtz | The MTZ file used for difference density in the rhofit run |
| Hit_00_00_000.pdb | The best position found for the ligand, in the best cluster, using the best dictionary for that cluster (this file is the same as best.pdb). |
| Hit_00_00_001.pdb | The 2nd best position found for the ligand in the best cluster (if more than one solution was found), using the best dictionary. |
| Hit_00_01_000.pdb | The best position found, in the best cluster, using the second-best dictionary |
| Hit_01_00_000.pdb | The best position found, in the second-best cluster, using the dictionary which worked best for the first cluster |
| best.cif | The CIF-format restraint dictionary corresponding to best.pdb. |
| cluster00.pdb, cluster01.pdb ... | The cluster files used by the fitter; the number on the file corresponds to the first of the two-digit numbers in the Hit name |
| dict00.cif, dict01.cif ... | The dictionaries used by the fitter; the number on the file corresponds to the second of the two-digit numbers in the Hit filename. Multiple dictionaries are produced only if you use -scanchirals; do 'diff input_dictionary.cif dict00.cif' to see which chiral centres have been flipped |
Smart OS, Womack, TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C and Bricogne G (2014) RHOFIT, version 1.2.4 Cambridge, United Kingdom: Global Phasing Ltd.
See separate page: History of improvements to rhofit
Please see: rhofit Wiki page
If rhofit crashes or produces poor results, please email buster-develop@globalphasing.com and tell us as much as you can about what you were doing when it crashed; if you can rerun the job with -td temp and send us temp.tar.gz, that would be perfect.
Please let us know if you would like any feature to be added to rhofit, by emailing buster-develop@globalphasing.com.