Direct use of weighted Quantum Chemical Energy for ligands in BUSTER refinement
QM advanced tutorial example:
raloxifene bound to 2qxs
Contents
Background
- This tutorial shows how to use more complex features of the -qm method, such as how to treat charged ligands and using a helper application for more accurate and expensive quantum calculations.
- For an introduction to the ideas behind the weighted-QM-energy as a restraint function for ligands see AutobusterLigandQMIntroduction, for an introduction to method see AutobusterLigandQMtutorialIntro2i0g.
- For this example we will use 2qxs:
- This is 1.7 Å resolution structure Estrogen Receptor Alpha with bound ligand raloxifene.
- rcsb page for 2qxs
- wikipedia page for raloxifene
- The structure is well determined with the two fold NCS and very clear density for both the ligands.
Starting files
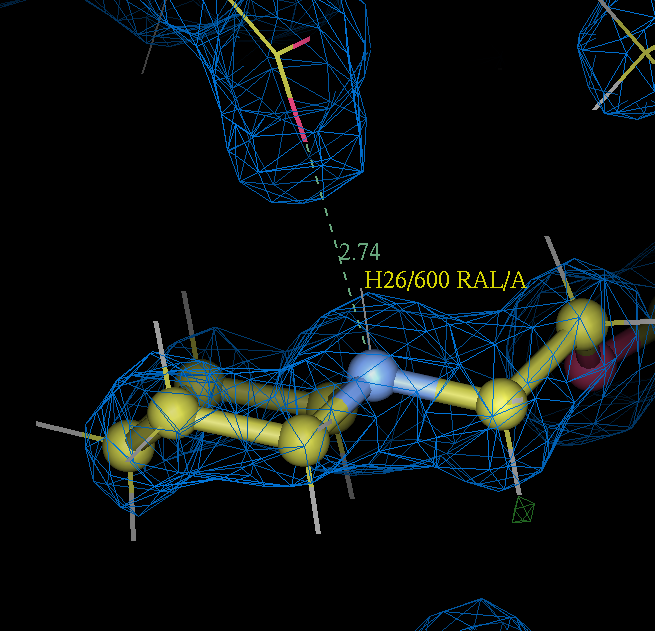
- raloxifene can be expected to be charged under crystallization conditions (pH 8.0) as the !pKa of 1- methyl piperidine is 10.0 ref.
- Furthermore in the refined 2qxs structure the raloxifene piperidine nitrogen atom forms as nice salt bridge with the side chain of ASP 351:
- So as a consequence a dictionary for a charged raloxifene RALplus.grade.cif was prepared:
- first grade_PDB_ligand RAL was used to prepare a dictionary for neutral raloxifene as described in ligand expo.
- the grade output PDB file was loaded into coot. In coot a pointer atom was defined 1 angstroms from N26 in the position expected for a piperidine proton (H26). This was used with an editor to produce a charged raloxifene pdb file RALplus_init.pdb that in turn was converted into mol2 format using babel. The charged raloxifene mol2 was then used as input to grade:
babel RALplus_init.pdb RALplus_init.mol2
grade -in RALplus_init.mol2 -charge 1 -ocif RALplus.cif -opdb RALplus.pdb
- The result is a dictionary with standard pdb atom names for raloxifene but with the additional proton. This dictionary was used with the hydrogenate utility to add hydrogen atoms to 2qxs for both protein and ligand atoms (see AutobusterLigandQMtutorialIntro2i0g for method). The resulting protein was refined and few clear improvements were made. The result is 2qxs_cleanup_refine.pdb.
(A) control refinement with grade dictionary
- first refine with using a normal grade representation of the ligand:
refine -p 2qxs_cleanup_refine.pdb \
-m 2qxs.mtz -autoncs -M TLSbasic -nbig 2 \
-l RALplus.grade.cif -d partA_d> partA.log
- note that -M TLSbasic uses TLS refinement for the first big cycle.
(B) refinement using RM1 for charged raloxifene
refine -p partA_d/refine.pdb \
-m 2qxs.mtz -autoncs -nbig 2 \
-TLS TLSfixcycALL=1 \
-l RALplus.grade.cif -qm RAL+1 \
-d partB_d> partB.log
- As each raloxifene has a charge +1 then turn on QM using -qm RAL+1. You can check that the charge is correctly specified by looking at the log file: partB_d/01-BUSTER/Cycle-1/LIST.html This has a section:
Have command line argument: '-qm RAL+1'
so will setup QM calculation
three-letter code decoded to be ='RAL' charge set to= 1
- Two big cycles are used -nbig 2
- TLS is used but the TLS parameters from the previous run are left unaltered by specifying -TLSfixcycALL=1 This means that coordinates are altered in both big cycles. This avoids using a big cycle to minimally readjust TLS parameters - important if using a slow expensive QM method.
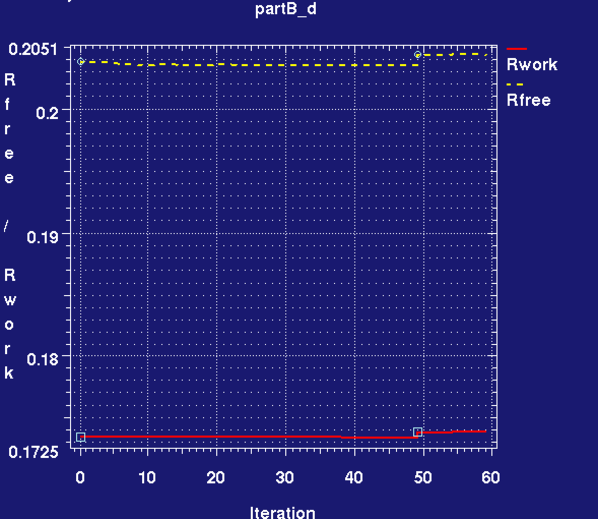
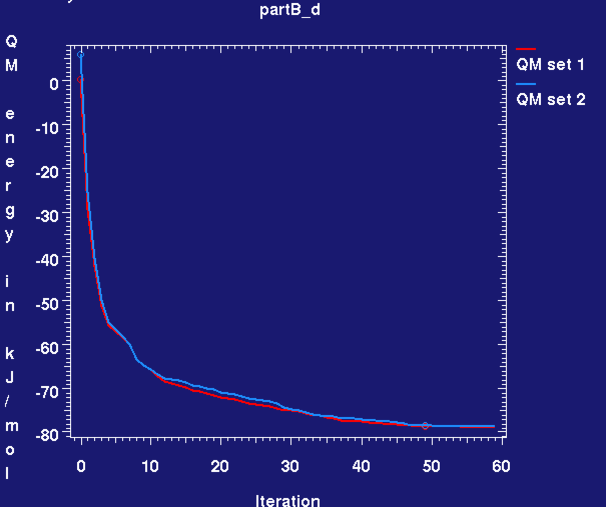
- during and after refinement the graph_autobuster_R and graph_autobuster_QM utilities can be used to monitor the change in R/Rfree and QM energy:
graph_autobuster_R -d partB_d -launch
graph_autobuster_QM -d partB_d -launch
 click to enlarge
click to enlarge click to enlarge
click to enlarge
- We can see that the Rwork and Rfree change little and there is a decrease in QM energy for both sites. This is due to a relaxation in the bond lengths and angles towards values that RM1 prefers with the overall ligand confirmation remaining pretty much unaltered, see analysis in part(D).
(C) refinement using GAMESS HF/3-21Gd with PCM water for charged raloxifene
- RM1 is an semi-empirical representation that is computationally cheaper but reasonably accurate. It is also possible to use more expensive quantum chemical methods by using an external helper application, See AutobusterLigandQMhelpers page for details on the helpers currently available.
- This will be illustrated here by using the GAMESS helper/program to represent the raloxifene. An ab initio HF method will be used with a small basis set, HF/3-21G(d) that performs reasonably well (see wikipedia page on basis sets)
- In order to reduce the effect of electrostatics the QM calculations are performed using the Polarizable Continuum Model (PCM) for water. See wikipedia page on PCM
- shell script partC.sh
- or type:
refine -p
refine -p partA_d/refine.pdb \
-m 2qxs.mtz -autoncs -nbig 2 \
-TLS TLSfixcycALL=1 \
-l RALplus.grade.cif -qm RAL+1 \
-qm_helper '/your/full/path/to/gelly_qm_helper_gamess.pl' \
-qm_method 'HF/3-21G(d)_pcm' \
-d partC_d> partC.log
- The -qm_helper command line option is used to specify the helper script to be used. Unless the script is on your path the full path must be used.
- The -qm_method option is used to specify the method to be used. The method is passed to the helper script by BUSTER when it is invoked - so valid values depend on what the script understands.
- note that -qm RAL+1 is used to specify that a QM set is to be used to treat each occurrence of a RAL residue in the structure (there are two in this structure). The +1 part of the option means that a charge of +1 is used for each RAL.
- TLS is used but fixed during the refine, see discussion in part (B)
- during and after refinement the graph_autobuster_R and graph_autobuster_QM utilities can be used to monitor the change in R/Rfree and QM energy - see part (B) for details.
(D) comparison of refinement results
- Using coot to compare the raloxifene ligand from part (A) refinement with grade dictionary representation (gold) with that part (C) where an ab initio representation was used shows that they are very similar.
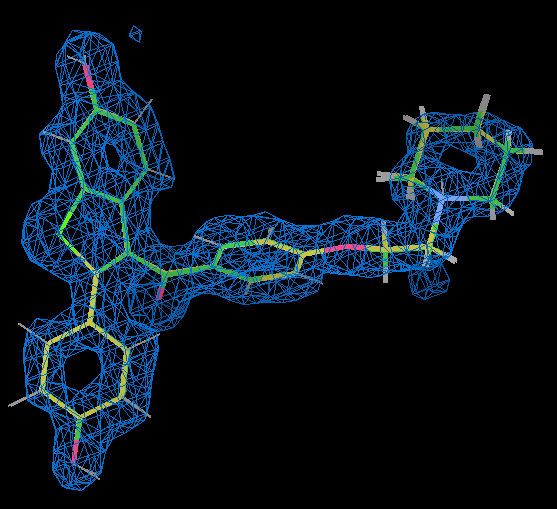
- This is also true of part (B) where RM1 is used for the raloxifene ligand. 2qxs is a 1.7 Å resolution and the ligand position is really well defined by the X-ray data as shown by the 2Fo-Fc density for it contoured at 1.5 sigma in the figure (the holes at the centre of the rings are a good indication of quality).
- A detailed comparison between the different refinements was made using the buster-report utility to speed calculations of ligand CC and mogul results. (buster-report is not yet released but will be soon).
- The differences in Rwork and Rfree are small, probably within noise but the lowest Rfree is with the ab initio method.
- The mogul rms Z score measures the bond lengths or angles found in comparison to that expected from CSD small molecule structure. A score of 1 indicates that most bonds are within 1 sigma of the expected value.
- As grade uses mogul to get target bond lengths and angles its Z scores are necessarily low. But for the QM representations mogul provides a useful indication of accuracy. It can be seen that the ab initio method produces a lower Z scores than the semiempirical.
- The GAMESS HF/3-21G(d) with PCM water run does take longer though. Using eight 2.80 GHz Xeon processors for the GAMESS calculations means the refinement takes around two hours.
- overall it can be concluded that using conventional restraints or either of the QM representations for the ligand yields good results.
(E) QM relaxation runs
- To gauge the internal strain of the ligand geometry optimization from the final position output in the refinement can be performed.
- For refinement in part (B) with RM1 the script partE1.sh can be used. or alternatively manually type
egrep "CRYST1|HETATM.*RAL A" partB_d/refine.pdb > partB_RAL_A.pdb
time gelly_refine \
-p partB_RAL_A.pdb \
-d partE1_d \
-o partE1_out.pdb" \
-l RALplus.grade.cif \
-qm RAL+1 \
-max 300 -glim 0.01 > partE1.log &
- So basically we grep out the first RAL ligand from the part B coordinates and then geometry refine this in isolation.
- The results of this run can be monitored during and after the run using the graph_autobuster_QM utility
graph_autobuster_QM -d partE1_d
... edited out ...
# final raw QM energies = 234.38 kJ/mol
# -qmzero not specified so report QM energies relative to first value read= 249.64
# big small iters QMener_01
# 0 0 0 0.00
# 0 300 300 -15.26
- This shows that the reported QM energy dropped by 15 kJ/mol to a final value of 234.38 kJ/mol
- using the final value as a basis line for part B:
graph_autobuster_QM -d partB_d -qmzero 234.38
... edited out ...
# big small iters QMener_01 QMener_02
... edited out ...
# 2 10 59 15.26 15.56
- So the QM strain energy according to RM1 is ~15 kJ/mol for both the A and B sites.
- Similarly for the ab initio run in part (C) a script: partE2.sh can be run using the GAMESS helper to relax the ligand.
- The conclusion of this is HF/3-21G(d) with PCM water gives a moderate strain energy of 10 kJ/mol for A site and 11 kJ/mol for B. This is
- coot can be used to examinine the motion associated with the ab initio relaxation by loading partC refine.pdb and refine.mtz together with partE2_out.pdb. This shows that ether type oxygen is slightly out of the phenyl plane in the refined result (gold and density) but this flattens on relaxation:
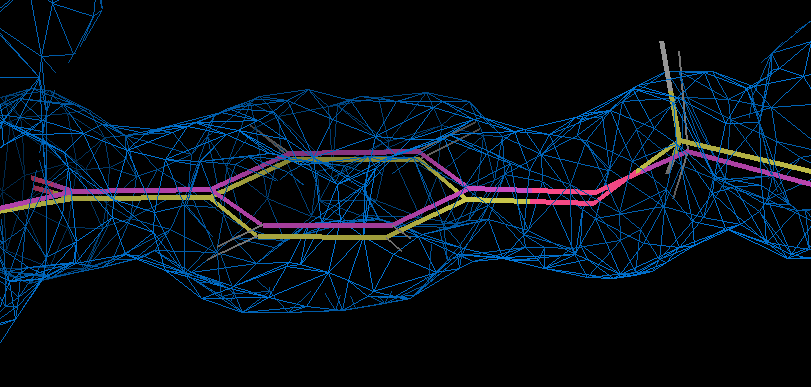
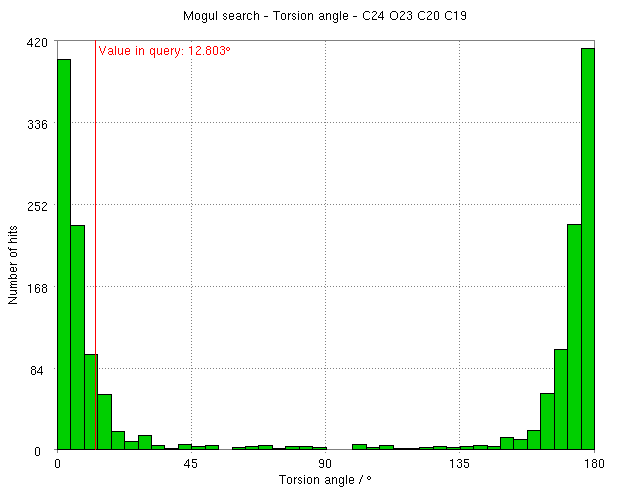
- The torsion angle C19-C20-O23-C24 changes from 12.8 degrees to 2.9 degrees on relaxation.
- mogul analysis of this torsion agrees that it is slightly strained and would prefer to be planar:
- In contrast, the RM1 result (E2) shows some limitations of this model. The major movement in the relaxation optimization is again in the ether type oxygen atom but this pushed out of the phenyl plane. The torsion angle changes from 21 degrees refined structure to -46 degees. The mogul result indicates this is wrong and the group has a preference to be coplanar.
Page by Oliver Smart from 13 May 2011. Any questions regarding our software or this wiki should be directed to buster-develop@globalphasing.com