| Attachments | |
|---|---|
| 1w50.pdb | 305K |
| summary.out | 4K |
| report2.png | 44K |
| report1.png | 184K |
| 4j0p.mtz | 1MB |
| post-refinement.png | 925K |
| 1w50.mtz | 1MB |
(revision 2.1, October 2024)
This tutorial represents a typical and straightforward case, involving two beta-secretase structures, that is run with default options. The reference structure is PDB id: 1W50 and the experimental data is taken from PDB id 4J0P.
| 4j0p.mtz | the structure factors as supplied by the pdb for 4j0p, converted to mtz format |
| 1w50.mtz | the structure factors as supplied by the pdb for 1w50, converted to mtz format |
| 1w50.pdb | the model coordinates as supplied by the pdb for 1w50 |
(A) Restraint dictionary generation
Prior to running Pipedream, we need to generate restraint dictionaries for the ligand(s) soaked into the experimental data and any prosthetic groups present in the structure in addition to the soaked ligand.
Pipedream WILL NOT generate dictionaries itself. Restraint dictionaries should be generated manually and validated before use in an automated procedure.
The experimental structure, 4j0p, was complexed with the compound N-[(4S)-2-amino-4-methyl-5,6-dihydro-4H-1,3-oxazin-4-yl]-4-fluorophenyl-5-cyanopyridine-2-carboxamide (PDB ligand id: 1H8) by soaking.
So, lets generate a restraint dictionary for the ligand, using Grade2.
Note that where the ligand is already present and classified by the PDB (as in this case), then a dictionary can be generated using grade2 -P:
grade2 -P 1H8
grade2 'c1(cnc(cc1)C(=O)Nc1cc([C@@]2(C)N=C(OCC2)N)c(cc1)F)C#N' -r LIG
Principle amongst the output files that grade2 produces are:
Now that we have prepared the restraint dictionary and have all the required input files, we are ready to run pipedream.
The one remaining check to be made is to ensure that the expected ligand binding site in the reference structure being used does not contain any other compounds. If so, they MUST be removed from the pdb file before use.
In this case, 1w50.pdb is a native structure and the binding site is clear, so no action is required. By default, Pipedream will automatically remove waters from the reference structure, so waters do not have to be removed manually.
Furthermore, there is a single bace monomer in the asymmetric unit, and hence a single binding site.
pipedream -hklin 4j0p.mtz -xyzin 1w50.pdb -hklref 1w50.mtz -rhofit 1H8.restraints.cif -d pipe &
Now, we will look at the output and the results.
The final R and R free after post-refinement (18.7% and 21.9%) look good. But the proof of the pudding is has the ligand been correctly fit and refined?
So let us look at the final output (and compare it with the deposited 4j0p structure):
visualise-geometry-coot postrefine-grade-LIG
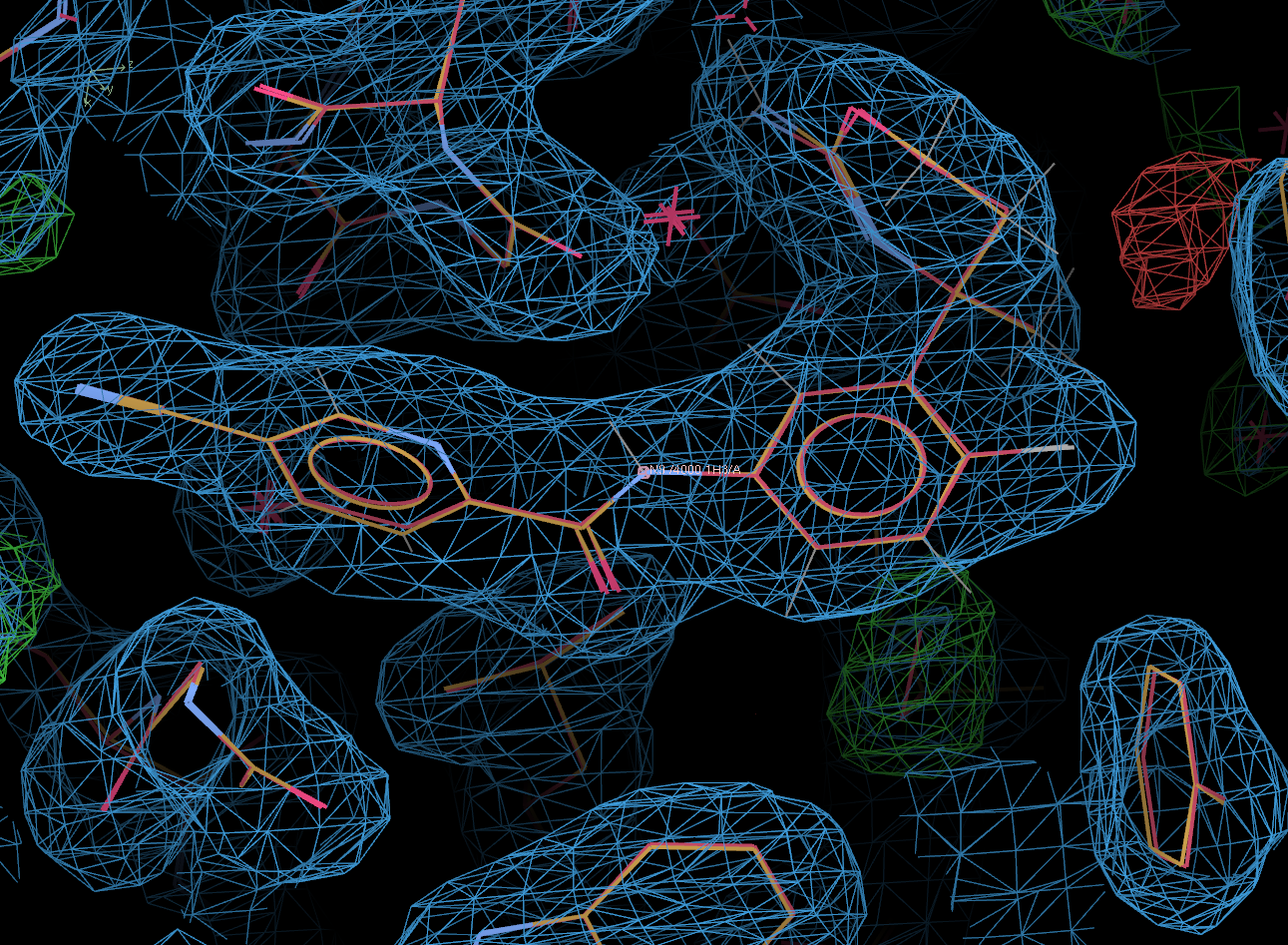
In the above, the original 4j0p model (including ligand) is shown in red.
Clearly, the solution found by Rhofit and post-refined is correct.
firefox report/index.html
or your favourite browser.
Look at the "Ligand report" tab for detailed analysis of all of the ligands present in the structure, in this case just the ligand fit by Rhofit.
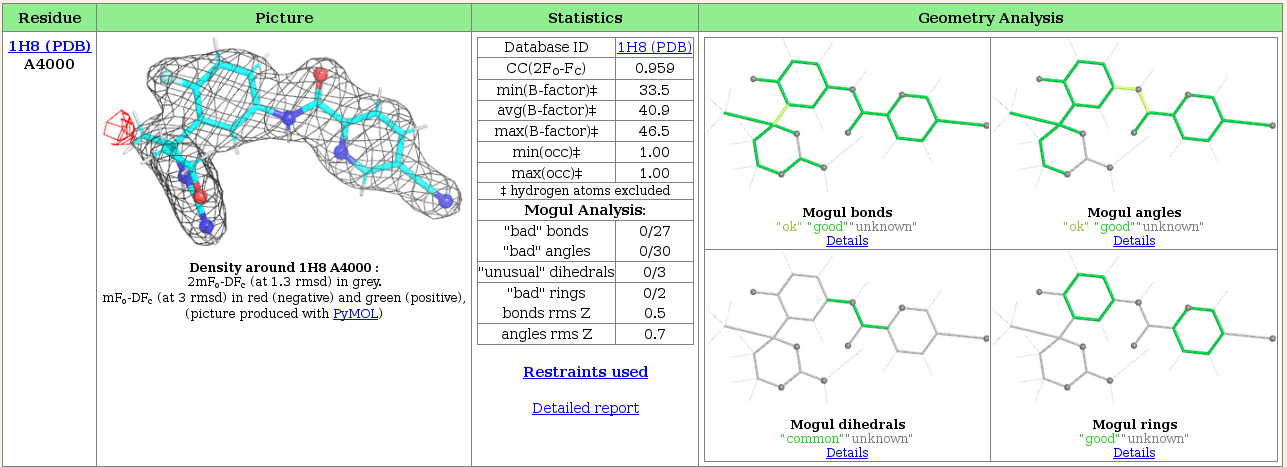
The centre section tabulates information to show how well the refined ligand agrees with the experimental data (the 2fofc correlation coefficient) and standard refinement parameters (Bfactors and occupancy). It also summarizes the Mogul analysis of the refined ligand. A more detailed analysis of the ligand geometry is shown in the "Geometry Analysis" pane on the right hand side, which, using colour coded pictures of the ligand shows Mogul's analysis of bond lengths and angles, dihedrals and rings. The colour coding used is:
In this case, the ligand has very good geometry.
Now look at the "molprobity analysis" tab. This shows the summary table of the Molprobity report for the final structure as well as the Ramachandran plot. A full molprobity chart listing per-residue violations is also available from this page.
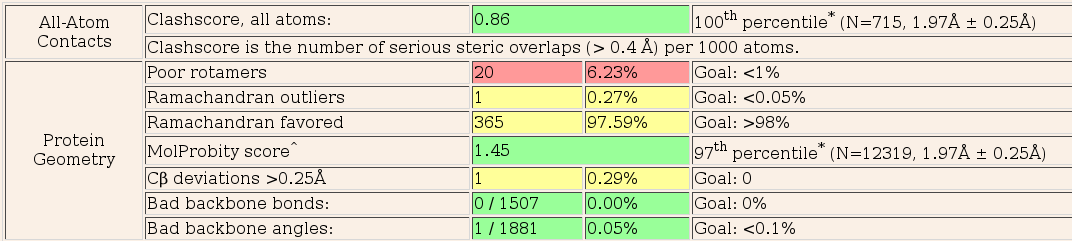
The molprobity analysis is pretty good but clearly there are still improvements that can be made. In particular, there are a number of residues with poor rotamers. These are not necessarily bad but may need looking at. Remember, the structure that is input into Pipedream has come from a different crystal from the experimental data and may also have been treated differently. There may well be conformational differences between the reference and experimental structures (due to a number of causes, not least ligand binding), that cannot be fully rationalised by refinement alone.
IMPORTANT: Pipedream is NOT designed to output a completed, fully refined, deposition ready structure! It may well be necessary to manually correct features of the structure in Coot and then carry out a further re-refinement.