UNDER CONSTRUCTION (throughout the workshop)
Content
Introduction
These pages are intended as a resource for the tutorials of our software at the International workshop, Joint CeBEM-CCP4 initiative,
April 9th-17th, 2013, Institut Pasteur Montevideo, Uruguay .
Much more introductory material is available for each program:
Local setup
All programs should be configured and setup correctly. You can test this by running
% process -h
% refine -h
and by pointing your browser (firefox) to http://localhost:8080/ (username/password = pasteur/sharp). If the SHARP server isn't running - try (re)starting it with
% $BDG_home/start.sh
If this doesn't work - please contact the tutors.
Helper applications
When looking at the results of the program (through your browser), there are links to graphical information like plots, PDB files, electron-density maps etc.
(When running autoSHARP through the CCP4i interface graphical results are presented as simple links instead!)
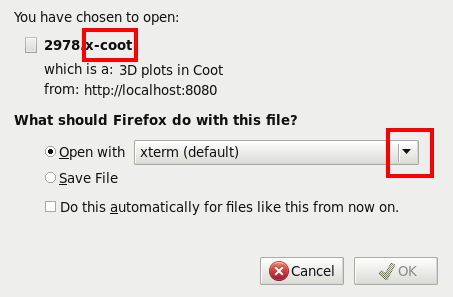
Those will be visualised through external applications like Coot - in the same way as you might look at PDF files with Acrobat when browsing the internet. Anyway, in order to get this going a little bit of work is required by the user the first time a specific type of result is encountered. You will see a pop-up appearing that looks similar to this:
We now need to tell it to use a specific helpers script that comes as part of our software installation - so the place where this helper script is residing on your computer might vary. It is a good idea to type (in a terminal):
% ls $BDG_home/bin/helpers.x-*
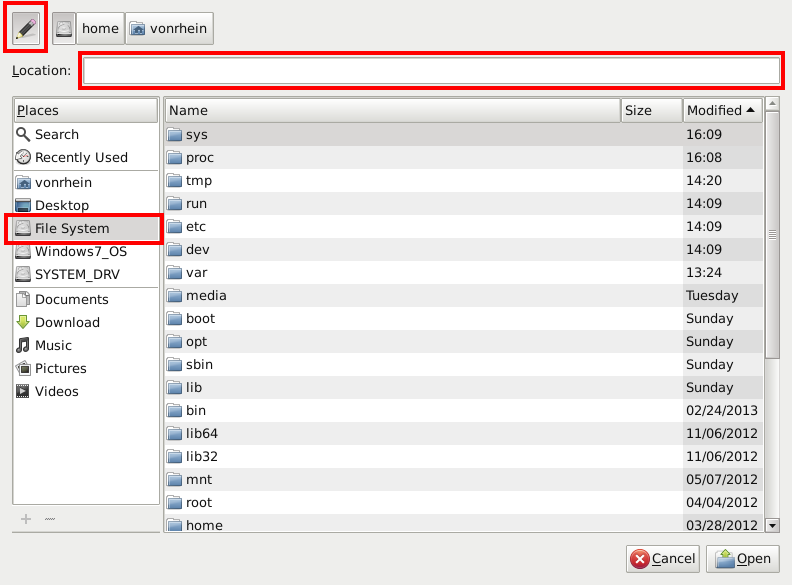
which will show the various helper script (for different type of files/results). Using the "Open with" item we can either browse to pick the right helper script or write the full path directly:
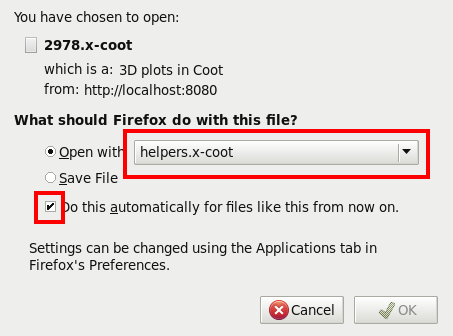
Finally, we want to use the same method every time - which will mean the correct helper application will pop up automaticaly the next time:
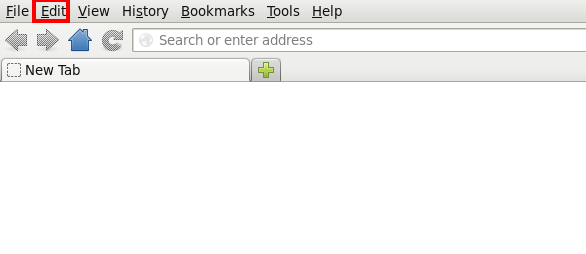
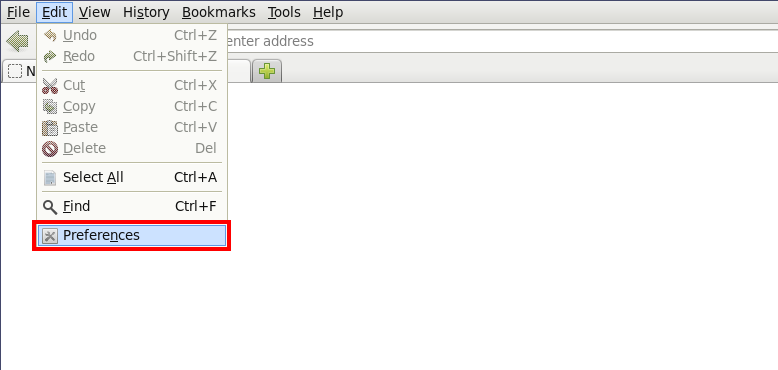
Sometimes it might become necessary to redo the steps above - but the browser doesn't ask the user to pick the correct helper application anymore. Then it becomes necessary to reset the handling within the browser, eg. in Firefox using the "Edit" ...
... "Preferences" ...
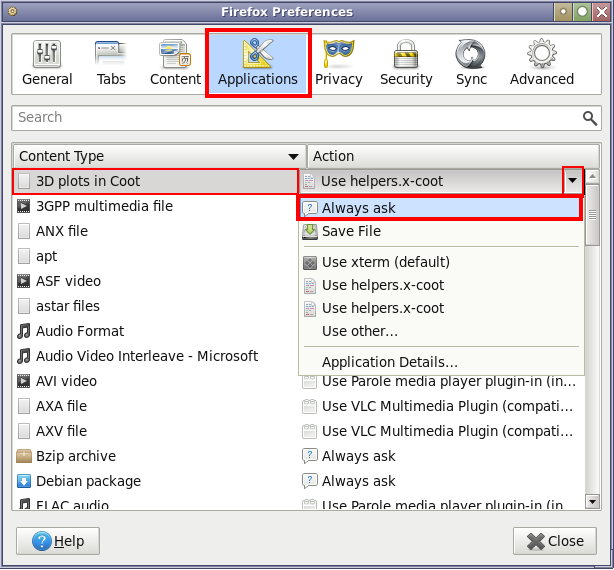
... "Applications":
After changing the action for the "3D plots in Coot" from "Use helpers.x-coot" back to the "Always ask", the above sequence of getting asked, browsing and selecting the appropriate helper application script can be done again.
1o22
This should be a reasonably fast run through all stages of data-processing (5 min), structure solution (15 min) and refinement (15 min) using a single example dataset (timings are for an i7-2720QM laptop with 8Gb of memory).
We're going to use the data for 1O22 from the JCSG. It is
- a 170 residue protein
- expressed with Se-MET (6 Met in sequence 1o22.pir)
- data collected at the peak wavelength (0.9778 A; 1o22_peak.sca)
- a fluorescence scan gave values of f'=-7 and f"=5.0
- detailed information available here
Hopefully, you managed to follow the autoPROC tutorial for processing the peak dataset (in which case you will have a file aimless.sca in the output directory). Alternatively, we also provide a reflection file below.
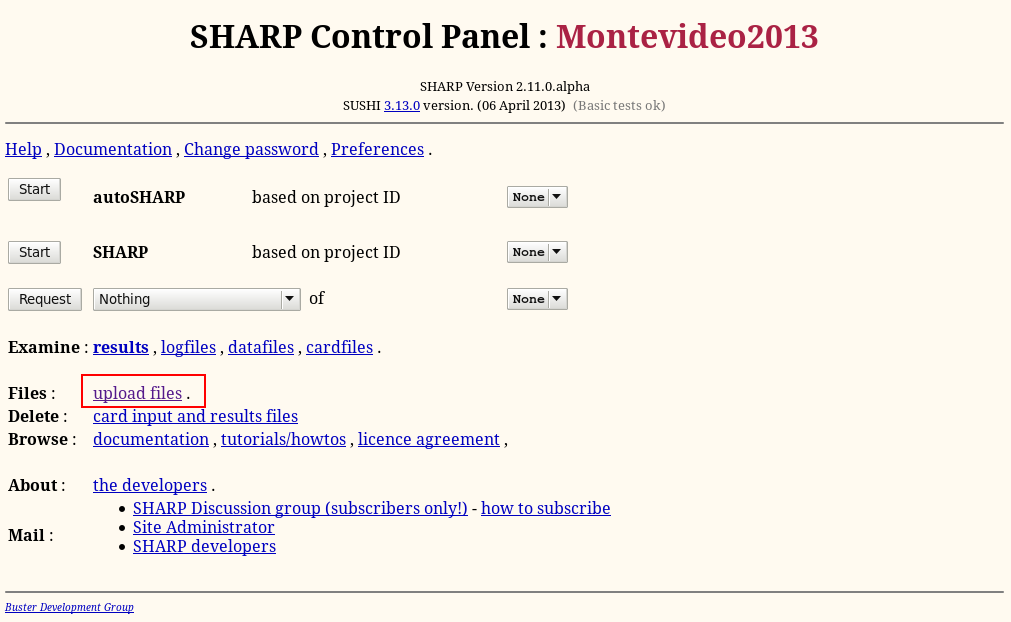
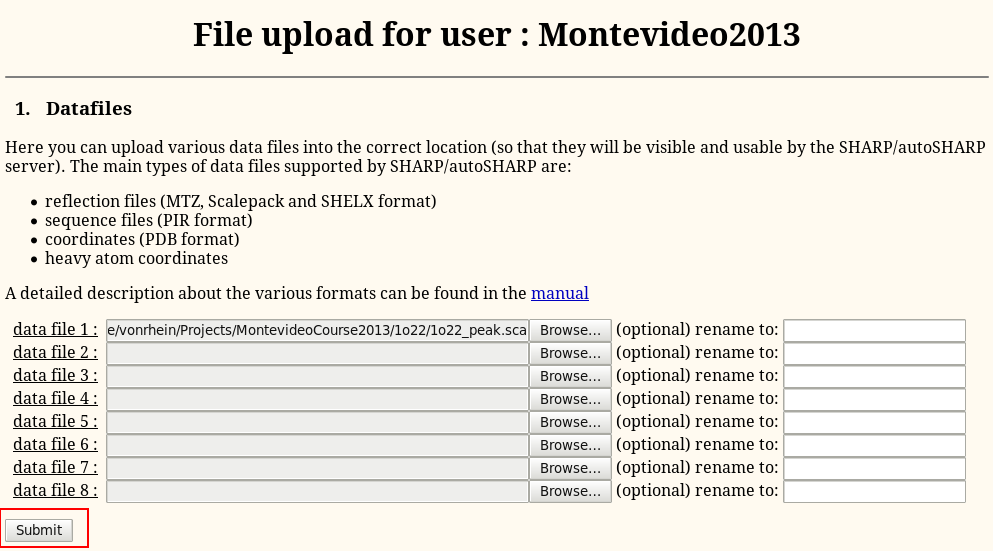
To get data into the program, start on the so-called "SHARP Control Panel" and the "Upload files" link:
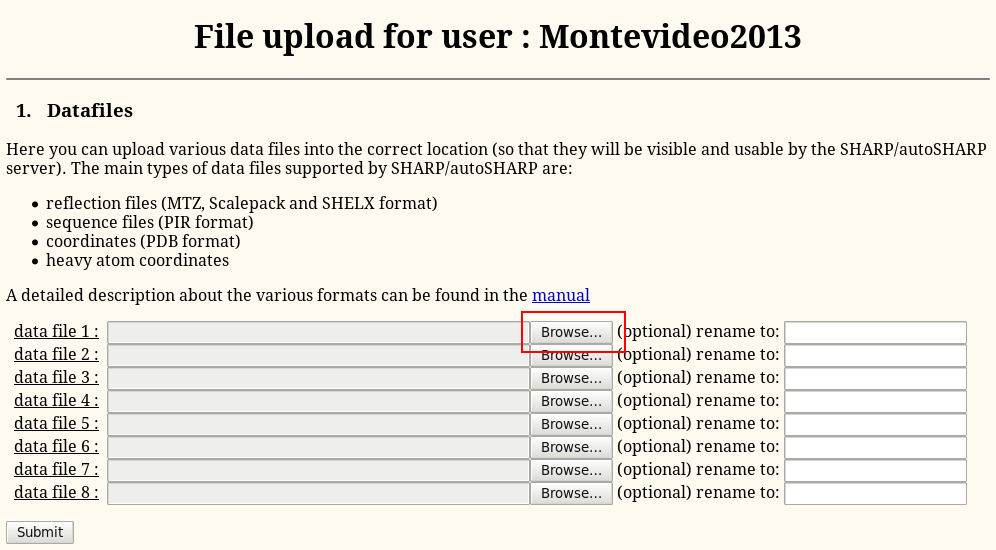
Then "Browse" to the file (MTZ, SCA, PIR, PDB etc - eg 1o22.pir and 1o22_peak.sca):
Finally, hit the "Submit" button
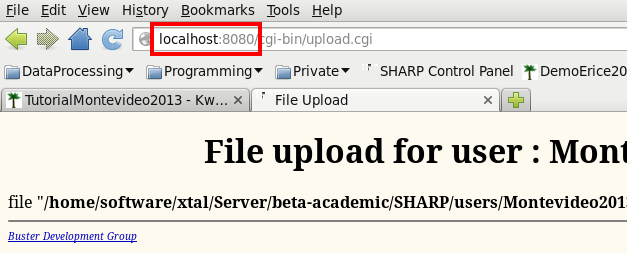
Remember: if you get lost you can always go back to the "SHARP Control Panel" using the URL http://localhost:8080/
SHARP/autoSHARP (experimental phasing and structure solution)
Please also have a look at the examples and tutorials on the SHARP/autoSHARP wiki.
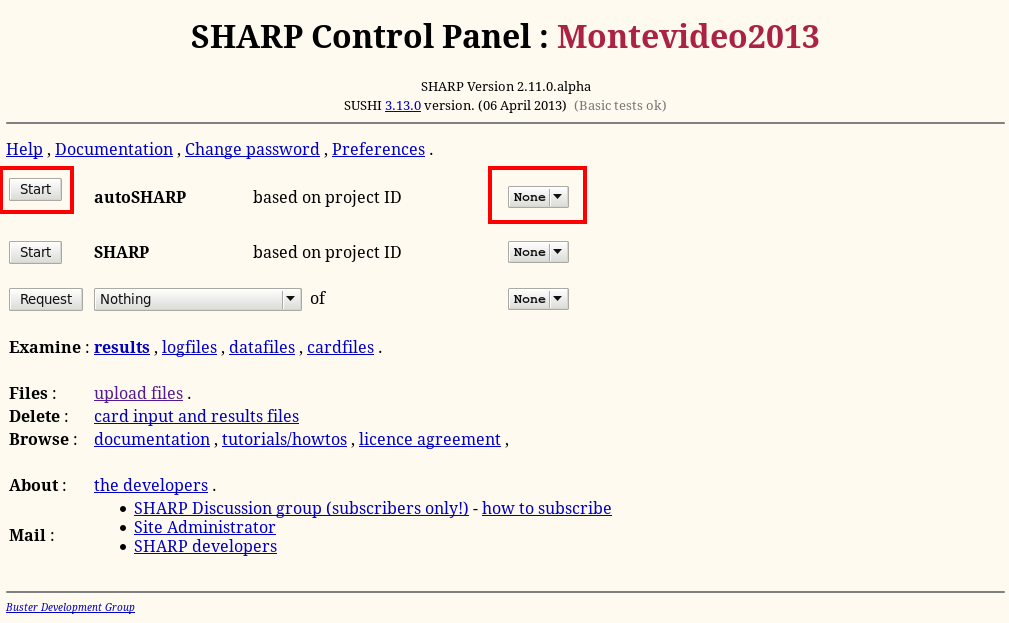
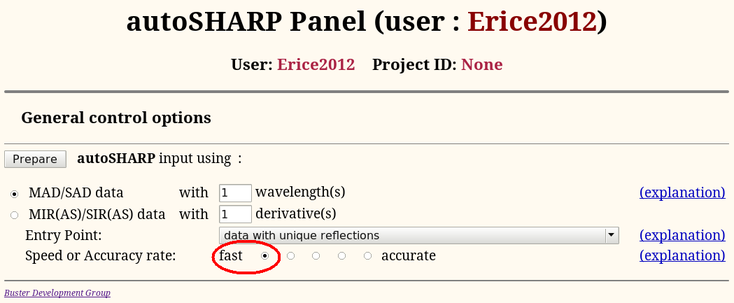
We can use the data just processed for solving the structure with autoSHARP (run through the Sushi http-interface). To start autoSHARP from the main "SHARP Control Panel", Start it afresh (ie. select "None" from the pull-down menu to the right:
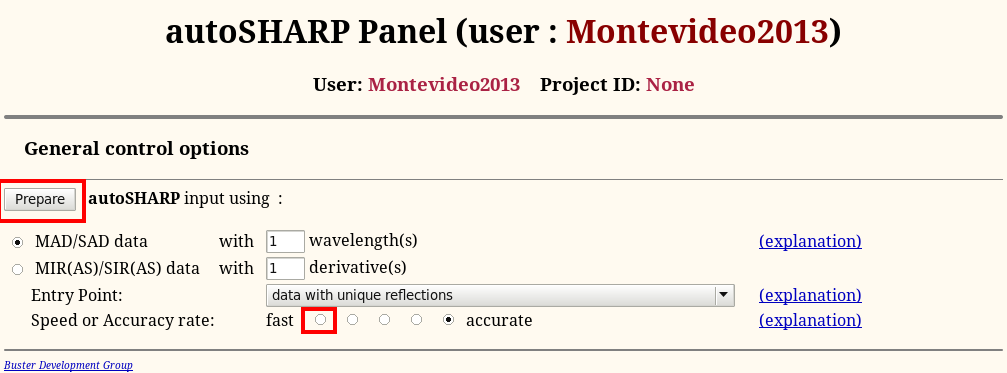
In order to have a quick run through the software during this tutorial, we will change the defaul mode to "fast":
In general, it is better to leave it at the default (accurate) to be more confident that a shortcut to achieve faster execution doesn't hinder successful structure solution.
Remember what we know about the project:
- searching for 6 Se atoms
- wavelength 0.9778 A
- f' and f" values of -7 and +5
- reflection file from autoPROC 1o22_peak.sca (you might need to still upload this into the program - see above; or use your own file resulting from re-processing the images)
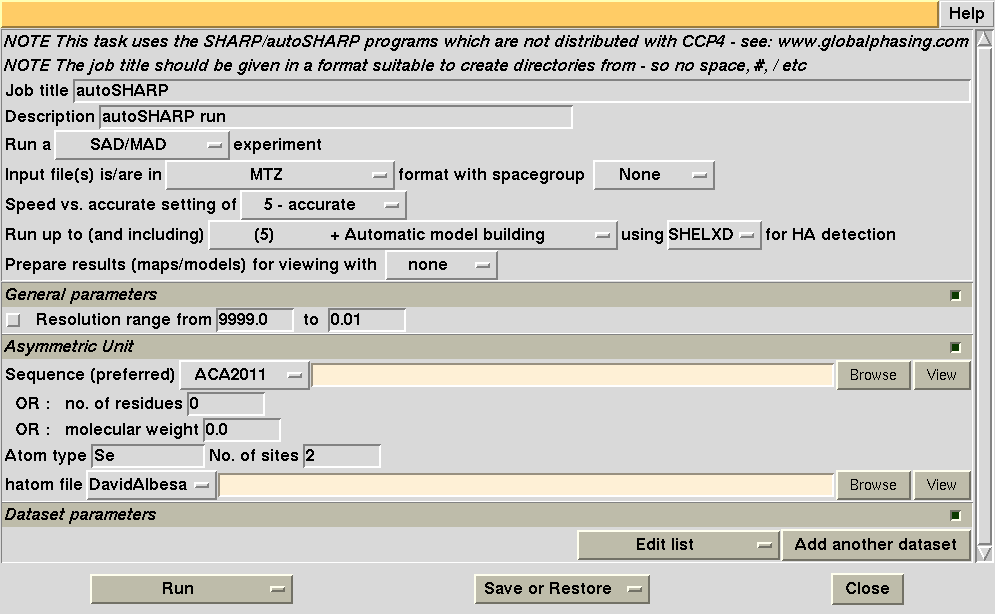
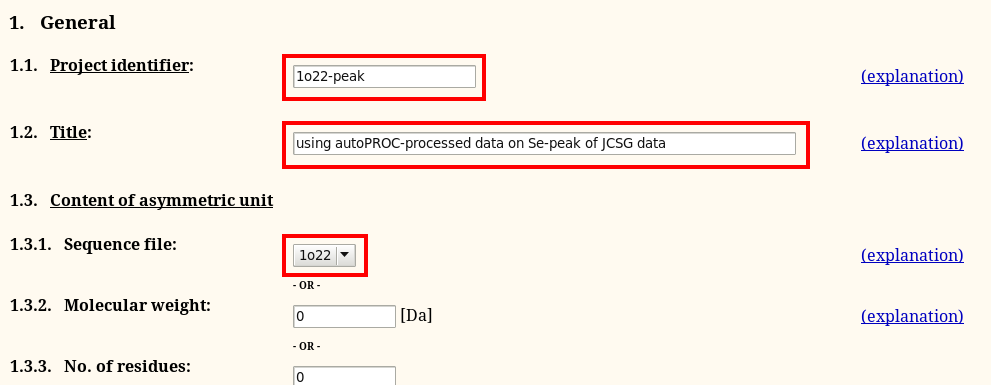
Filling in this information will look like this: first we need to give our job a meaningful and short identifier (without spaces or any "funny" characters). We also want to select the sequence file 1o22.pir (containing sequence of a monomer):
The remainder is relatively easy too - make sure you give the number of expected sites per monomer too:
the final step - just submit the calculation
and continue to the main autoSHARP logfile:
Rember that you need to "reload" that page from time to time:
After about 10 minutes we should have
- found the heavy atom sites (with SHELXC/D)
- refined these, calculated phases (with SHARP)
- density modified the experimental map (with SOLOMON)
- built an initial map (using ARP/wARP, REFMAC)
This model has 145 out of 170 residues buit with R/Rfree = 0.219/0.259 :
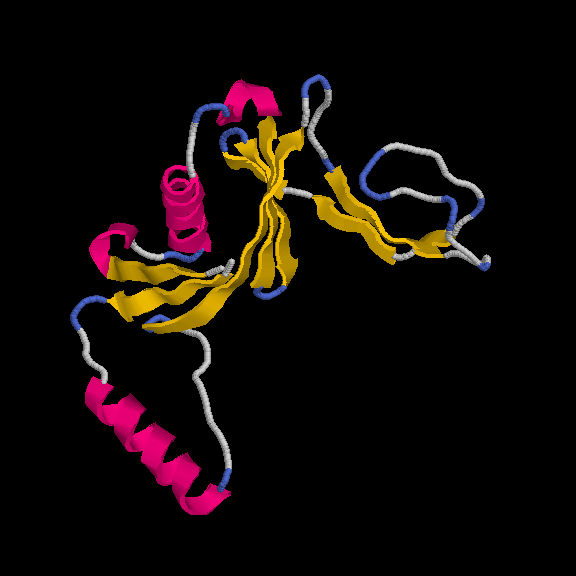 Remember, the deposited model contains 149 residues - so we are basically finishing with a complete model.
Remember, the deposited model contains 149 residues - so we are basically finishing with a complete model.
own data
This is the fun bit - working with your own data. You should be able to get very far all on your own. But if there are any questions: just let us know - also during coffee breaks etc!
SHARP/autoSHARP (experimental phasing and structure solution)
Make sure you have your sequence in a simple ASCII file and that you have a very good idea about your fluorescence scan (for anomalous signal data). You can get data files into the SHARP server (the httpd) through the 'Upload' mechanism on the main page.
There is an alternative (but less well tested and supported) interface to autoSHARP within CCP4i:
which leads you to
Original page: April 2013, Clemens Vonrhein



{kind=link}