The -forcefield refine option allows the use of a force field to represent a ligand. It uses the procedures developed for the Direct use of weighted Quantum Chemical Energy for ligands in BUSTER refinement. But instead of using a quantum chemical representation for the ligand a molecular mechanics force field is used. Initial work has concentrated on providing helpers for the MMFF94 force field (that is widely used and well regarded). Force fields offer a number of advantages compared to quantum methods:
The OpenEye implementation of MMFF94 includes extensions enabling the forcefield to represent compounds containing selenium (details) or boron (details). MMFF94 is used in the OpenEye AFITT automated ligand fitter.
$BDG_home/setup_local.sh $BDG_home/setup_local.csh
refine -p model.pdb -m data.mtz -d 01 -l grade-LIG.cif -forcefield LIG > 01.log
refine -p model.pdb -m data.mtz -d rund -l grade-LIG.cif -forcefield LIG > run.log
run/01-BUSTER/Cycle-1/LIST.html
QM_HELPER_LOG: forcefield.pl invoking OpenEye Helper with /mnt/scratch_fs1/osmart/autobuster/Server/scripts/qm-mm-helpers/OpenEye.pl 0 1 AUTO
QM_HELPER_LOG:
QM_HELPER_LOG: gelly helper script to run OpenEye helpers for force field or QM energy/gradient calculation
QM_HELPER_LOG: Please obtain helpers for OpenEye http://www.eyesopen.com/
QM_HELPER_LOG: helper script location /mnt/scratch_fs1/osmart/autobuster/Server/scripts/qm-mm-helpers/OpenEye.pl
QM_HELPER_LOG: picked up charge=0 multip=1 method=AUTO from command line
QM_HELPER_LOG: Open Eye executable used: /home/osmart/2013/09/OpenEyeHelper/openeye/bin//buster_helper_mmff
QM_HELPER_LOG: :jGf:
QM_HELPER_LOG: :jGDDDDf:
QM_HELPER_LOG: ,fDDDGjLDDDf, BUSTER HELPER MMFF
QM_HELPER_LOG: ,fDDLt: :iLDDL;
QM_HELPER_LOG: ;fDLt: :tfDG;
QM_HELPER_LOG: ,jft: ,ijfffji, :iff
QM_HELPER_LOG: .jGDDDDDDDDDGt.
QM_HELPER_LOG: ;GDDGt:''':tDDDG,
QM_HELPER_LOG: .DDDG: :GDDG.
QM_HELPER_LOG: ;DDDj tDDDi
QM_HELPER_LOG: ,DDDf fDDD, Copyright (c) 2013
QM_HELPER_LOG: LDDDt. .fDDDj OpenEye Scientific Software, Inc.
QM_HELPER_LOG: .tDDDDfjtjfDDDGt
QM_HELPER_LOG: :ifGDDDDDGfi. Version: 2.3.0.4
QM_HELPER_LOG: .:::. Built: 20130710
QM_HELPER_LOG: ...................... OEChem version: 1.9.2 20130710
QM_HELPER_LOG: DDDDDDDDDDDDDDDDDDDDDD Platform: Ubuntu-12.04-g++4.6-x64
QM_HELPER_LOG: DDDDDDDDDDDDDDDDDDDDDD
QM_HELPER_LOG:
QM_HELPER_LOG:
QM_HELPER_LOG:
QM_HELPER_LOG: Input ISM: c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
QM energy for QMset 1 picked up as 316.47988
QM_HELPER_LOG: Input ISM: c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
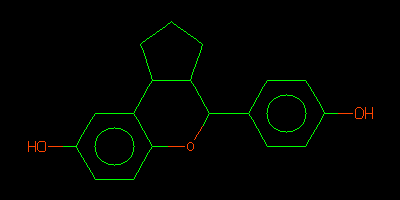
egrep ISM rund/01-BUSTER/Cycle-1/LIST.html | head -1 | sed -e 's/.*ISM..//' > check.smi obabel check.smi -Ocheck.svg convert check.svg check.png
Currently the -forcefield option and helpers have the major limitation that ligands must be chemically complete with all hydrogen atoms. This limitation has consequences for handling both covalent and incomplete ligands.
Ligands that are incomplete (have missing atoms) will not be handled properly at present. Any problem should be apparent by checking the SMILES string reported by the helper ISM line. It should be possible to fix this limitation in a future release.
The current procedures will not properly handle covalently-bound ligands including proteins containing modified amino acids or nucleic acids with modified bases. Any problem should be apparent by checking the SMILES string reported by the helper ISM line. It should be possible to fix this limitation in a future release.
The current procedure does not handle normal alternate positions in ligands. It is possible to treat alternates using a workaround, if you would like details contact buster-develop@globalphasing.com
It should be possible to fix this limitation in a future release.
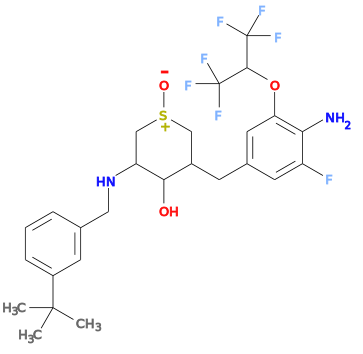
Currently grade will incorrectly set the type for S=O bonds to "single". This will result in an incorrect force field setup, this will be shown by checking the SMILES string reported by the helper ISM line. For instance for the 1YU ligand from structure 4lxm the ISM SMILES produces a structure:
| Back to Ligand QM/force field top index page |
Page by Oliver Smart Nov 2013, modified June 2014 Any questions regarding our software or this wiki should be directed to buster-develop@globalphasing.com