BUSTER -forcefield option
Introductory tutorial on 2i0g.
Contents
Background
Starting Files
- Use the (recently release) fetch_PDB tool to download the PDB and structure factors for 2i0g, and converting the SF to mtz format.
fetch_PDB 2i0g
- prepare a grade dictionary for I0G by
grade_PDB_ligand I0G
(A) Preparation: add hydrogen atoms to the ligand(s)
- Please note that it is essential to add hydrogen atoms to the ligand for the -forcefield option to work, see AutobusterLigandForcefield for details.
- run command:
hydrogenate -p 2i0g/2i0g.pdb -l I0G.grade_PDB_ligand.cif
-ligonly -zero -o 2i0g_addhI0G.pdb
(B) Refine with -forcefield I0G -l I0G.grade_PDB_ligand.cif
- First add hydrogen atoms to the ligand.
- The refine option -forcefield I0G will use the MMFF94 force field in place of conventional restraints for every copy of I0G in the protein:
time refine \
-p 2i0g_addhI0G.pdb \
-m 2i0g/2i0g.mtz \
-autoncs \
-l I0G.grade_PDB_ligand.cif \
-forcefield I0G \
-d partBff -report > partBff.log &
- Since 2i0g has 2-fold NCS the -autoncs option is used. LSSR restraints will be used for both the protein and the I0G ligand.
- Note that a CIF restraint dictionary must still be specified for the ligand.
- To check that the MMFF94 force field is being used for the ligand, use firefox (or other browser) to examine the initial log file:
firefox partBff/01-BUSTER/Cycle-1/LIST.html
- Scroll down until you see a section:
QM_HELPER_LOG: forcefield.pl invoking OpenEye Helper with /mnt/software/GPhL/ALL_snapshot_20131118/scripts/qm-mm-helpers/OpenEye.pl 0 1 AUTO
QM_HELPER_LOG:
QM_HELPER_LOG: gelly helper script to run OpenEye helpers for force field or QM energy/gradient calculation
QM_HELPER_LOG: Please obtain helpers for OpenEye http://www.eyesopen.com/
QM_HELPER_LOG: helper script location /mnt/software/GPhL/ALL_snapshot_20131118/scripts/qm-mm-helpers/OpenEye.pl
QM_HELPER_LOG: picked up charge=0 multip=1 method=AUTO from command line
QM_HELPER_LOG: Open Eye executable used: /software/OpenEye/openeye/bin/buster_helper_mmff
QM_HELPER_LOG: :jGf:
QM_HELPER_LOG: :jGDDDDf:
QM_HELPER_LOG: ,fDDDGjLDDDf, BUSTER HELPER MMFF
QM_HELPER_LOG: ,fDDLt: :iLDDL;
QM_HELPER_LOG: ;fDLt: :tfDG;
QM_HELPER_LOG: ,jft: ,ijfffji, :iff
QM_HELPER_LOG: .jGDDDDDDDDDGt.
QM_HELPER_LOG: ;GDDGt:''':tDDDG,
QM_HELPER_LOG: .DDDG: :GDDG.
QM_HELPER_LOG: ;DDDj tDDDi
QM_HELPER_LOG: ,DDDf fDDD, Copyright (c) 2013
QM_HELPER_LOG: LDDDt. .fDDDj OpenEye Scientific Software, Inc.
QM_HELPER_LOG: .tDDDDfjtjfDDDGt
QM_HELPER_LOG: :ifGDDDDDGfi. Version: 2.3.0.4
QM_HELPER_LOG: .:::. Built: 20130710
QM_HELPER_LOG: ...................... OEChem version: 1.9.2 20130710
QM_HELPER_LOG: DDDDDDDDDDDDDDDDDDDDDD Platform: Ubuntu-12.04-g++4.6-x64
QM_HELPER_LOG: DDDDDDDDDDDDDDDDDDDDDD
QM_HELPER_LOG:
QM_HELPER_LOG:
QM_HELPER_LOG:
QM_HELPER_LOG: Input ISM: c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
QM energy for QMset 1 picked up as 870.70907
- This shows that the MMFF94 force field is being used to assess the energy of the chain A copy of I0G. It's very important to verify that the SMILES string is correct. The SMILES is reported in the line(s):
QM_HELPER_LOG: Input ISM: c1cc(ccc1C2C3CCCC3c4cc(ccc4O2)O)O
- It is very strongly recommended that you check the chemistry by producing a 2-D diagram. How to do this is described in detail in the Checking section of AutobusterLigandForceField wiki page.
- While the refinement is running you can check how the force field energy in kcal/mol for the two copies of I0G changes by:
graph_autobuster_QM -d partBff -k -qmzero 0 -launch
- This produces a plotmtv graph which looks like this (here the run has only completed 60 small cycles):
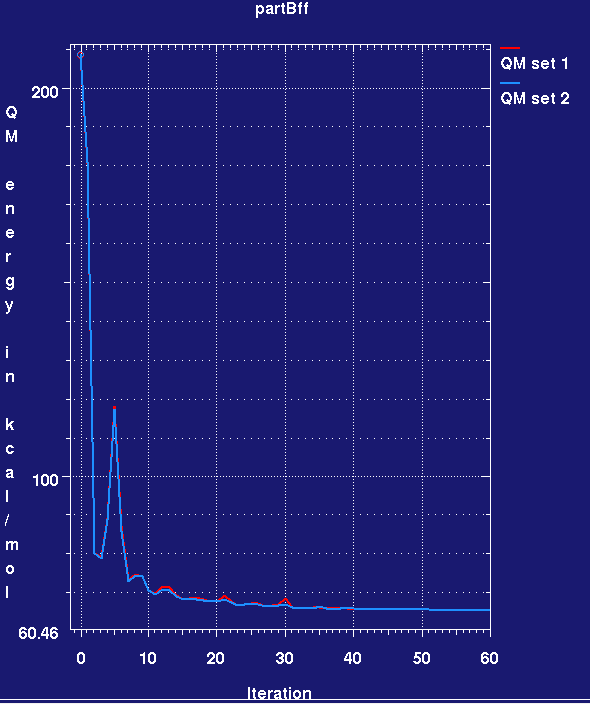
- Note that the MMFF94 force field energy for the two ligands rapidly drops in the refinement and then remains steady. Also note that we cannot yet assess ligand strain as we do not know the force field energy for a reference low-energy conformation of the ligand.
- Once partB has completed examine the IOG ligand geometry.
visualise-geometry-coot partBff
- In COOT also load 2i0g.pdb for comparison.
- Use COOT "Go to atom" to find IOG A 1.

- Here the MMFF94-refined ligand is shown in orange, and compared with the original PDB conformation in green. The 2Fo - Fc density is contoured at 1.3 RMSD and the difference density at 3 RMSD.
- In terms of fitting the macromolecular data the changes are small and the fit to the map is the same.
- However, if you use buster-report and Mogul to examine the bond lengths and angles for the QM-refined I0G in comparison to the original I0G from the PDB file, many fewer deviations are found (see analysis below).
- Looking at the Rwork/Rfree graph for the completed partB job, the refinement produces a nice drop in Rfree by about 1.2%. Is this due to the use of QM for the ligand? Of course it isn't! - the difference is the result of running a long refinement with BUSTER, particularly with -autoncs. We will show this by running a control refinement in part (C).
(C) Control refine with dictionary
- As a control do the same refine job but leave out the -qm option so that the I0G geometry will be handled by the GRADE dictionary:
time refine \
-p 2i0g_addhI0G.pdb \
-m 2i0g/2i0g.mtz \
-autoncs \
-l I0G.grade_PDB_ligand.cif \
-d partC -report > partC.log &
- In addition a MapOnly run can provide analysis of the starting PDB file:
time refine \
-p 2i0g_addhI0G.pdb \
-m 2i0g/2i0g.mtz \
-M MapOnly \
-l I0G.grade_PDB_ligand.cif \
-d MapOnly -report > MapOnly.log &
(D) Analysis comparing the Force Field, QM and dictionary refine jobs
- It should be noted that the BUSTER refinement improves the Rfree and the MolProbity overall score by a similar amount with all three representations. The differences are small. This is to be expected: all three refinements use reasonable ligand representations.
- Now examine the metrics for I0G A 1 (the ligand from the A chain).
- Note that the map correlation coefficient is lower in all three refinement compared with the original PDB entry. It is the job of a good ligand representation (restraints, force field, or QM) to keep chemistry sensible and so avoid over-fitting the noisy low-resolution density. The Mogul bond metrics for the PDB ligand are not good suggesting that a poor refinement dictionary was used.
- The MMFF94 results indicate that it performs slightly better than RM1. A higher ligand correlation coefficient is produced together with a better bond angle metric as compared to CSD expectations from Mogul.
(E) Assessing ligand strain local conformation energy and motions for the refinement using MMFF94
- In part (B) we saw how it is possible to monitor the force field energy for the ligand during a refinement relative to its initial position. However, what we really want to assess is the local conformational strain energy of the ligand. To do this we want to find what the force field energy of the ligand in its refined position is with respect to the minimum energy conformation of the ligand. Normally this can be found by doing a "energy minimization" of the ligand in isolation from its position in the refined complex.
- First extract the ligand coordinates for the A site. This can be done with an editor or using the egrep command. N.B.: BUSTER requires that all PDB files have a CRYST1 line:
egrep "I0G A|CRYST1" partBff/refine.pdb > partBff.IOGA.pdb
- Then use gelly_refine to geometry optimize the ligand:
gelly_refine -p partBff.IOGA.pdb \
-l I0G.grade_PDB_ligand.cif -forcefield I0G \
-d partEff -o partEff_out.pdb \
-max 1000 -glim 0.01 -jiggle_xyz 0.1 > partEff.log &
- The run uses a dictionary and -forcefield IOG
- It also increases the default maximum number of steps and reduces the convergence limit: this is useful to ensure that a minimum is actually reached. The -jiggle_xyz 0.1 argument jiggles the input coordinates by a small amount: this is useful to ensure the optimizer does not get stuck in a saddle point.
- The command graph_autobuster_QM can be used to find the MMFF94 force field energy of the ligand in the optimization.
graph_autobuster_QM -d partEff -k -q 0
final raw QM energies = 263.77 kJ/mol
- So the MMFF94 force field energy for the ligand is 263.77 kJ/mol
- This energy can be used as a reference point to find the local conformational strain energies for ligand in partB refine:
graph_autobuster_QM -d partBff -qmzero 263.77 -k
# -qmzero specified so report QM energies relative to 263.77
# kcal set scale energies by 0.238845896627496
...
# 5 63 357 1.73 1.73
- This means the upper limit of the local conformational strain energy for the ligand is 1.7 kcal/mol N.B. recent work has produced an improved method for estimating ligand conformational strain that produces lower values. This will be scripted and released soon
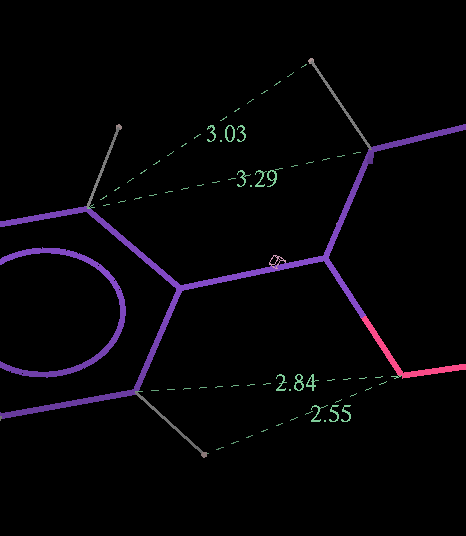
- The optimized coordinates for the ligand partE_out.pdb (purple) can be compared to the refined ligand (orange) using COOT:

- It can be seen the relaxation is principally a rotation of the phenyl ring. The 2Fo-Fc density in the refined protein clearly defines the orientation of the ring. Relaxation of the ring within the protein would cause it to have a short contact with Leu 339.
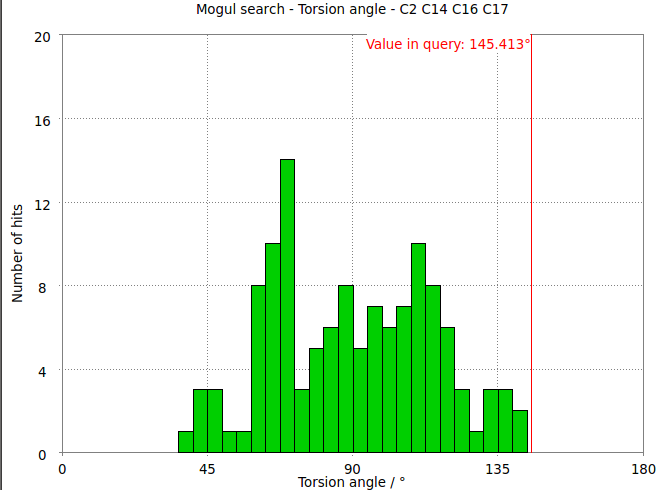
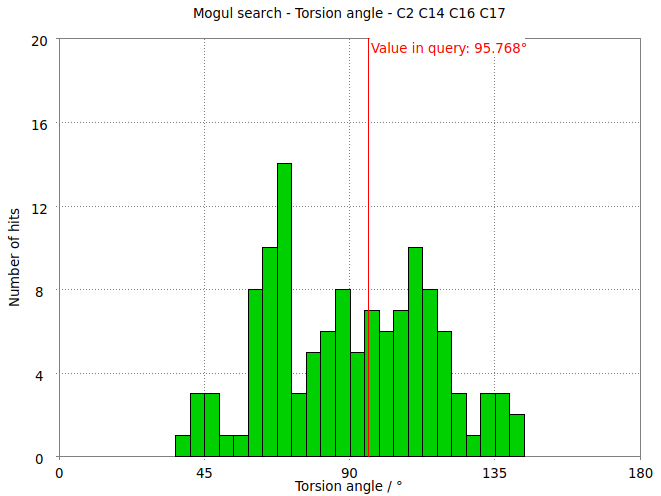
- The torsion angle C2-C14-C16-C17 measures the rotation of the phenyl ring. This torsion angle changes from -145.6 degs in the refined structure (partB) to -97.8 degs.
- It is interesting to compare these results with the Mogul distribution of similar torsion angles from CSD small molecule structures. In the refined structure the ligand phenyl torsion angle is regarded as "rare" by buster-report as it is on the edge of the expected torsion angle from related CSD structures :
- But relaxation with the MMFF94 force field allows shifts the torsion angle into the range to be expected for small molecule structures.
- The relaxation relieves short contacts between the phenyl ring and rest of the ligands.
Page by Oliver Smart and Ian Tickle 18 Nov 2013. Any questions regarding our software or this wiki should be directed to buster-develop@globalphasing.com