Comparing Grade2 and EH99 Restraints for amino acid side chains¶
Introduction¶
Grade2 produces restraints for ligands that are based on information from the Cambridge Structural Database (CSD) of small molecule structures where possible. This chapter examines the compatibility of Grade2 restraints with the EH99 (Engh and Huber, 2006) restraints for amino acids that are used in BUSTER refinements for proteins. EH99 restraints were obtained by an analysis of CSD structures using different methods when the CSD contained just over 200 thousand structures compared to today when it has over 1.2 million structures (Statistics on the Cambridge Structural Database).
It is shown how Grade2 bond and angle restraint ideal values are consistent with EH99 values. Furthermore, the agreement between the sigma values is examined with the conclusion that it is necessary to scale up Grade2 bond and angle restraint sigmas for complete consistency with EH99.
We thank a Grade Web Server user for raising this matter.
Method¶
Grade2 restraint dictionaries were produced for 17 common proteinogenic
amino acids (excluding GLY, ALA and PRO). The bond and
bond angle restraints were compared to the EH99
(Engh and Huber, 2006)
restraints for each of the side chains. Data were analyzed
using Jupyter Notebook with
matplotlib and
scipy.stats
Results¶
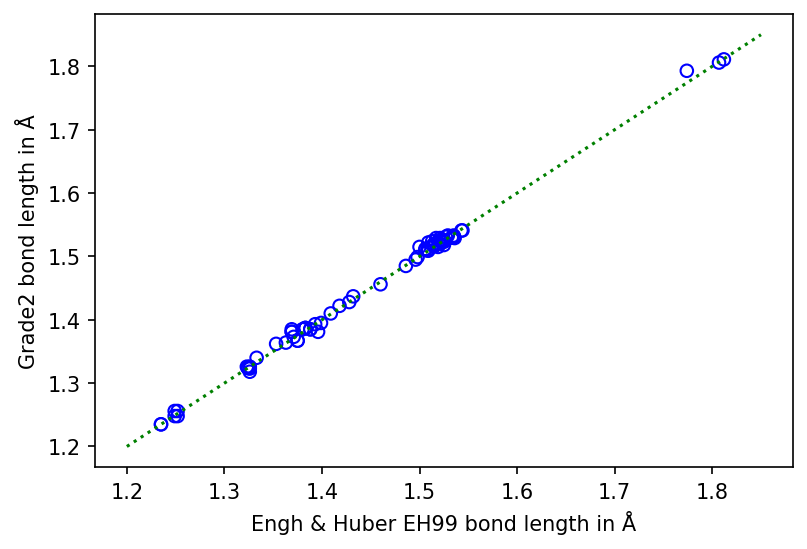
Comparison between the ideal bond lengths of EH99 and Grade2 restraints for amino acid side chains. Blue circles are used to mark each ideal bond length. The green dotted line marks equality.¶
As shown in the figure above, the ideal bond lengths are directly related (Pearson's r (78) = 1.00, p < .001). The root mean squared difference (rmsD) between the ideal bond lengths for the two sets of restraints is 0.006Å. This can be compared to an mean bond sigma value of 0.022Å for EH99 and 0.013Å for Grade2. The rmsD between the two sets is therefore approximately half the lower of the mean sigma values. It can be concluded that ideal bond lengths of Grade2 reproduce EH99 values consistently, as would be expected given they are both based on CSD structural information.
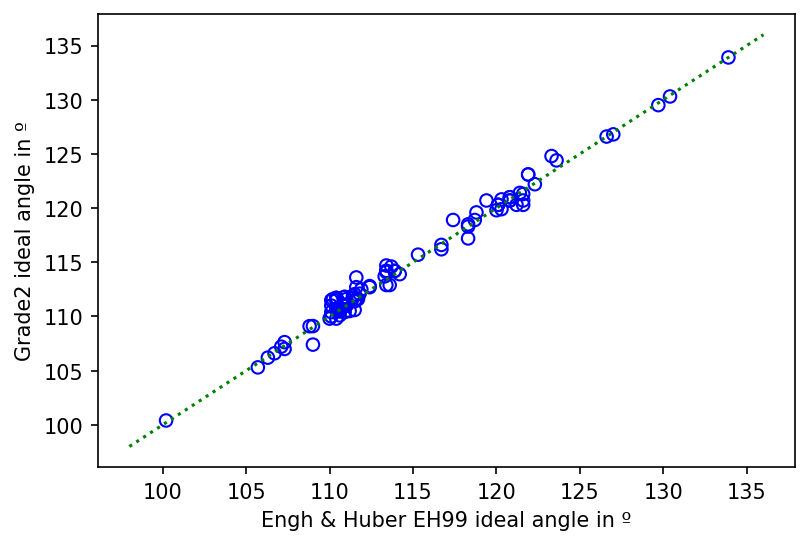
Comparison between the ideal angles in EH99 and Grade2 restraints for amino acid side chains.¶
As shown in the figure above, the ideal bond angle in EH99 and Grade2 are also directly related (Pearson's r (104) = .99, p < .001). The rmsD between the ideal bond angles for the two sets of restraints is 0.7º. This can be compared to an mean EH99 bond angle sigma value of 1.7º and 1.4º for Grade2. Once again, the rmsD between the two sets is therefore approximately half the lower of the mean sigma values. It can be concluded that ideal bond angles also agree well.
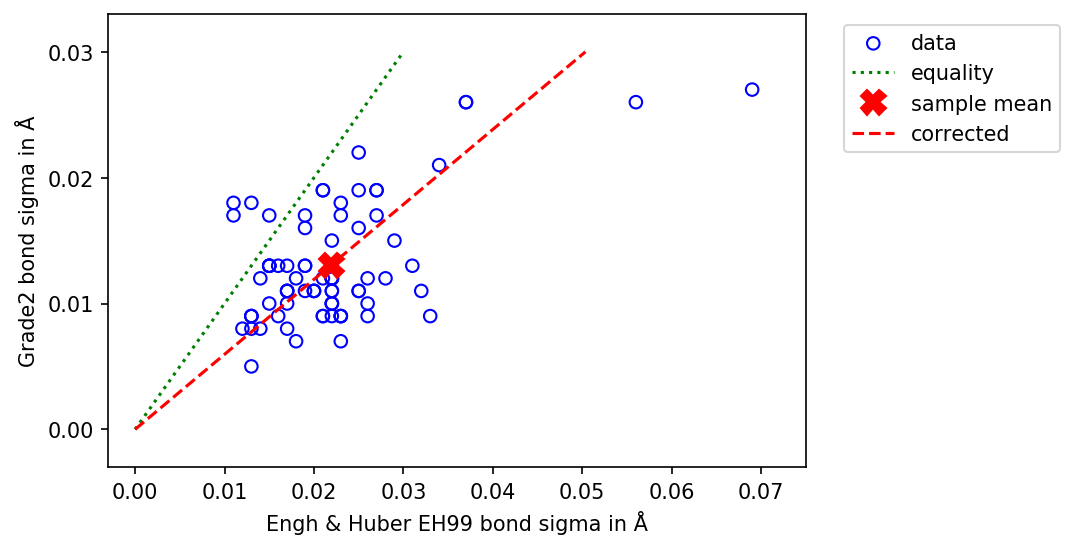
Comparison between the bond sigma of EH99 and Grade2 restraints for amino acid side chains.¶
Comparing the sigma values for bond lengths shows a moderate positive correlation (Pearson's r (78) = .58, p < .001). The Grade2 value for sigma are smaller than corresponding EH99 value in 95% of bonds. The mean sigma value for this set of bonds is 0.013Å for Grade2 compared to 0.022Å for EH99. The ratio of the two means is 1.68.
In a similar fashion the sigma values for bond angles (below) show a moderate positive correlation (Pearson's r (105) = .53, p < .001). The Grade2 angle sigma 1.4º is smaller than than the mean EH99 angle sigma 1.7º by a factor of 1.25.
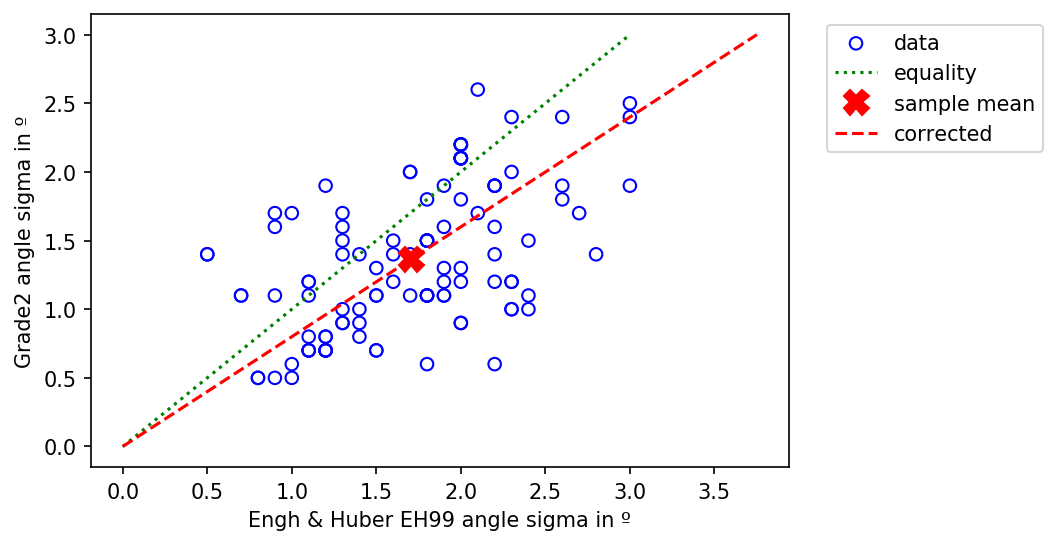
Comparison between the bond angle sigma of EH99 and Grade2 restraints for amino acid side chains.¶
A further investigation was conducted to find out why a tighter distribution for both bond lengths and bond angles is produced by Grade2 compared to EH99 values. Grade2 uses a Mogul option to only analyze CSD structures with an Rfactor <=5%. When this filter was disabled, the mean amino acid bond sigma increased by a factor of 1.63 and the mean angle sigma by 1.32. As these values are comparable to the difference between Grade2 and EH99, the major difference between Grade2 and EH99 is likely to be because Grade2 data is based on a selection of the highest resolution CSD structures rather than a wider set.
The --eh99_sigma_correction option¶
Grade2 from release 1.5.0 includes a
command line option --eh99_sigma_correction that scales up sigma values
for bonds and angles to
match the mean sigma values of the EH99 amino restraints:
The sigma values for bonds, that do not involve hydrogen atoms, are increased by a factor 1.68.
The sigma values for bond angles, that do not involve hydrogen atoms, are increased by a factor 1.25.
The --eh99_sigma_correction option can be used for refinements
where EH99 restraints
are used for protein residues, for instance in BUSTER.
If it is not used then ligand bond and angles will be restrained
to ideal values more strongly in comparison to protein residues
(for most users this will not be a problem).
The option should not be used if Grade2 restraints
are to be used with CCP4 restraints for amino acids. Comparing Grade2
and CCP4 restraints for amino acid bonds shows they have comparable
mean sigmas for both bond length and bond angles (Unpublished data).