See also:
"buster-discuss" site:globalphasing.com
A good start is to always run
checkdeps -n
to exercise various programs from the BUSTER package and check some basic functionality.
We also distribute a test script refine_test.sh (in $BDG_home/samples/autobuster directory) which runs a small refinement, using the options that we would recommend for regularising a structure and generating a new map after you've done a bit of rebuilding in coot. Further information on how to run it and on how long it takes to run on several machines we have tested it on can be found on the the BusterShortRefineTest page.
Please see the dedicated page on interpreting BUSTER output.
For relatively high-resolution X-ray data, the use of TLS can make difference density clearer at the end of a refinement. Once modelling (to take account of difference density) is starting to become frustrating, running BUSTER with -M TLSbasic sometimes improves the density and helps during model adjustments. Once you've started using TLS, it's probably best to keep using it for the rest of the refinement.
For low-resolution data and models where there is a well-understood domain structure, TLS may well be useful from the early stages.
The BUSTER geometry function incorporates a very strong BCORREL term which restrains the B-factor difference between bonded atoms, so neighbouring atoms are restrained to having similar B-factors. This type of restraint seems to work well even at resolution ranges where one would have used grouped B-factors refinement previously.
At very high resolutions you may want to consider using individual ADP refinement via the -M ADP macro.
The "unhappy atom" category uses the information from the analysis of restraint outliers (so-called geometry screen output), that sums the geometry function contribution of each atom for each restraint (bond, angle, torsion, contact term etc). The sorted list of those atoms contributing the most to the overall geometry function value provide the basis for this "unhappy atom" classification.
The list of these atoms can serve as an indication of problematic hotspots that would require some visual inspection against the 2mFo-DFc and mFo-DFc (difference) maps.
If the parameter AnalyseVoids is set to 'yes' (which is the default) the bulk solvent correction in the final cycle attempts to account for hydrophobic voids, i.e. regions within the model that are neither occupied by atoms nor by bulk solvent. We have found that this is often useful in removing patches of negative difference density from, for example, the interior of helix bundles. If you find that the change is for the worse on one of your structures, try running with refine AnalyseVoids=no ....
We provide some macros specifically for short BUSTER refinements after some model rebuilding with coot: see AutoBusterShortRunMacros page for details.
We provide a simple tool to generate appropriate commands (in Gelly-syntax) describing the most common classes of occupancy refinement:
The analysis is based on some basic assumptions, like
Running e.g.
% pdb2occ -p your.pdb -o occref.gelly % refine -p your.pdb -Gelly occref.gelly ...
will first analyse the existing PDB file (creating the text file occref.gelly), which is subsequently used in refinement. The text file can also be used as a starting template for defining more complicated situations. For further information and a full example please see AutoBusterLigandAndOccRef.
You can use the parameter BusterFreeFlagValue to select an explicit test-set flag:
% refine BusterFreeFlagValue=0 ...
It defaults to '0' to follow the usual CCP4 selection - see also AutoBusterFreeRFlag.
The MTZcolumns page aims to explain what each column means and how they could/should be used.
Please note that for PDB deposition we provide PDBx/mmCIF files that should be used directly as-is: the use of conversoin or extraction tools (like PDB_EXTRACT, SF_CONVERT etc) should be avoided if at all possible!
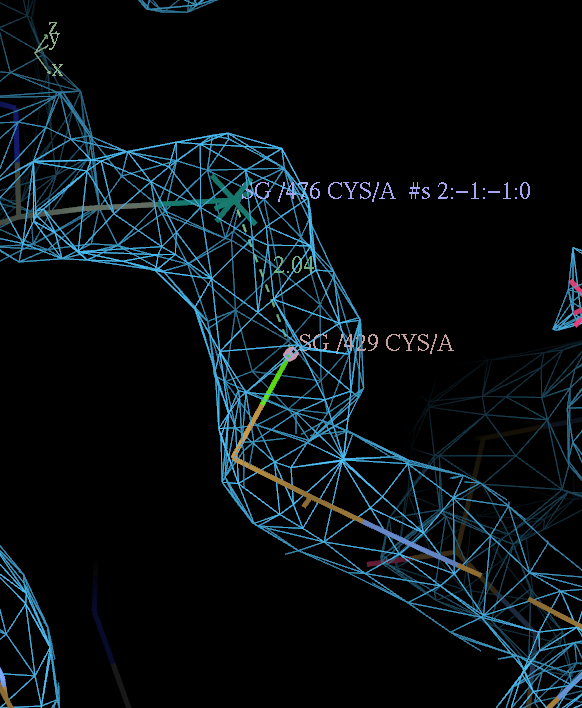
This can be done by adding a SSBOND card before the CRYST1 line in the input PDB file. The card must have correct column spacing.
SSBOND 1 CYS A 429 CYS A 476 1555 4445 2.04
key: c nnnn d mmmm oooooo
change 'c' and 'nnnn' to the chain id residue number of the identity copy CYS
'd' and 'mmmm' to the chain id residue number of the symmetry related CYS
leave 'oooooo' alone (provided it is not 1555 BUSTER will work out the correct symmetry operator).
PDB entry 4J0U provides an example with correct column spacing and a pretty disulfide across a symmetry contact:
If that's all you have, then the general answer is no; it's entirely possible for a PDB file arising from a sufficiently badly driven refinement process to lack critical information about the chemistry of the ligand. But if you also have a good idea as to what the chemistry is, have a look at HydrogenatePDBwithOpenBabel
The order in which settings can be changed from the defaults when running BUSTER:
1. installation-wide
This means a .autoBUSTER file in the same directory as the command (e.g. 'refine'). Or in more practical terms, these files should be located in
% dir=`which refine` % ls -l `dirname $dir`/.autoBUSTER
% set dir=`which refine` % ls -l `dirname $dir`/.autoBUSTER
2. user-wide
A file in the users home directory, i.e.
$HOME/.autoBUSTER
3. directory-wide
A file in the current directory (where the command, eg. 'refine' is being issued):
./.autoBUSTER
4. environment
Setting environemental variables in the usual shell-dependent way, eg.
% MyParameter="XYZ" % export MyParameter % refine ...
% setenv MyParameter "XYZ" % refine ...
5. command-line
% refine MyParameter="XYZ" ...
or
% refine -M MyMacro ...
where a file MyMacro.macro contains something like
# my fancy stuff MyParameter="XYZ"
and the directory containing that MyMacro.macro file being specified in the autoBUSTER_MacroDirs environmental variable.
Remember, the command-line is read in order, so
% refine MyParameter="ABC" -M MyMacro ...
will have a different effect to
% refine -M MyMacro MyParameter="ABC" ...
There is a big advantage of just using command-line arguments of the form par="value": the complete command-line is written into the header of the final refine.pdb file - so you always know exactly how it was run.
Please see the dedicated page Optimal number of processors for BUSTER.
You should be able to set a specific Perl binary at installation time via
% env BDG_perl=/where/ever/bin/perl ./GPhL_BUSTER_snapshot_20210224_install.sh
to have this binary set within the BUSTER installation (instead of taking the location of "perl" from the PATH at installation time).
If you need to use another Perl version than the one found when BUSTER was installed, you can do the following:
% env BDG_perl=/opt/myperl/bin/perl KeepFromEnv_BDG_perl=yes hydrogenate ...
If this works, you could add the following lines to your $BDG_home/setup_local.(c)sh files:
# setup_local.csh setenv BDG_perl /opt/myperl/bin/perl setenv KeepFromEnv_BDG_perl yes
and
# setup_local.sh export BDG_perl=/opt/myperl/bin/perl export KeepFromEnv_BDG_perl=yes
Remember to re-source the setup file again via
% source $BDG_home/setup_local.csh # tcsh/csh - or - % . $BDG_home/setup_local.sh # sh/bash/zsh/ksh/dash
The core refinement part of BUSTER is a fairly complicated multi-threaded program, and as such needs rather a lot of stack space. If you get error messages of the form
OMP abort: Unable to set worker thread stack size to 4194304 bytes
you can try some of these solutions:
If any MakeTNT application (MakeTNT, PDB2TNT, MDL2TNT, MOL22TNT, EditTNT, EditREFMAC or MakeLINK) fails to run please see MakeTNT applications do not run page for advice.
BUSTER requires that the file system it's running on supports the creation of UNIX sockets - the Linux and Darwin local filesystems and NFS do this, but AFS does not. If you see this message, try running BUSTER in a directory on a local filesystem.
BusterFAQ page. Please address problems, corrections and clarifications to buster-develop@globalphasing.com