| Attachments | |
|---|---|
| visualise-geometry-coot_02.png | 204K |
| R-Rfree_small.png | 56K |
| 1o22_truncate-unique.mtz | 884K |
| model_1o22-peak-try2.5_wARP_48.0pc.pdb | 99K |
| visualise-geometry-coot_01.png | 41K |
Content
These pages are intended as a resource for the tutorials of our software at the International workshop, Joint CeBEM-CCP4 initiative, April 9th-17th, 2013, Institut Pasteur Montevideo, Uruguay .
Much more introductory material is available for each program:
All programs should be configured and setup correctly. You can test this by running
% refine -h
which will already give you some basic help about BUSTER. You should also have a BUSTER reference card and please have a look at the information available on the BUSTER wiki.
Especially the introductory tutorial can be very useful.
We're going to use the data for 1O22 from the JCSG. It has originally been refined against 2.0 A data to an R/Rfree of 0.182/0.238 (see paper).
Hopefully, you managed to follow the autoPROC tutorial for processing the peak dataset (in which case you will have a file truncate-unique.mtz in the output directory). Alternatively, we also provide a reflection file: 1o22_truncate-unique.mtz.
You should also have an initial model from running this peak data through autoSHARP - alternatively, we provide the result of such an autoSHARP run: model_1o22-peak-try2.5_wARP_48.0pc.pdb.
What would be a good strategy to start refinement with BUSTER of that structure? We first need to set the spacegroup in the MTZ file to the correct one (which was the one we could solve the structure in - see your SHARP/autoSHARP result). One could do that within eg the CCP4i - or quickly on the command-line with
% echo "SYMM P43212" | mtzutils hklin 1o22_truncate-unique.mtz hklout 1o22_truncate-unique_P43212.mtz
The new reflection (MTZ) file will now be used for a run of BUSTER using 0 a reflection/MTZ file 0 a model/PDB file
% refine -m 1o22_truncate-unique_P43212.mtz -p model_1o22-peak-try2.5_wARP_48.0pc.pdb -d buster_01 | tee buster_01.lis
Since this is the first BUSTER refinement, you might see a few warning messages at the beginning: unless they are very severe, refinement should be able to improve the model enough that these will disappear.
To look at the resulting model and maps, you could use our tool specifically designed to help detecting any problematic regions in the model:
% cd buster_01 % visualise-geometry-coot
This allows to easily navigate through the most problematic residues (in terms of geometry):
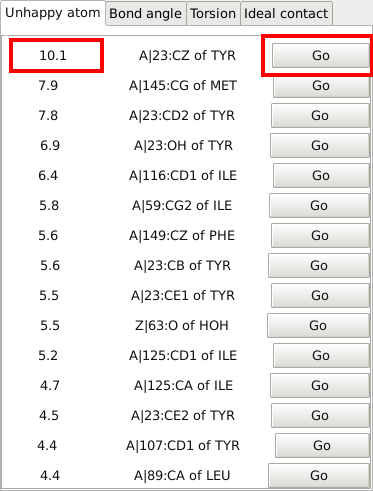 |
The score on the left shows the severity of the geometric problem - and with the right button you will jump to the problematic atom/residue:
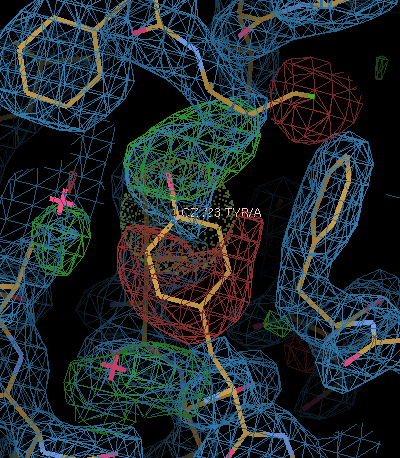 |
Or use Coot directly with "coot --pdb refine.pdb --auto refine.mtz". Of course, you should also use the other Coot tools for analysing problems (Ramachandran plot, difference-density peaks, unmodelled blobs etc).
This will allow you to easily correct the few geometry problems still present in the model (side-chain placement, missing residues, misplaced waters). After saving the corrected model, you can run a quick BUSTER refinement directly from within Coot or run again from the command-line with
% refine -m 1o22_truncate-unique_P43212.mtz -p buster_01/refine-coot-0.pdb -d buster_02 | tee buster_02.lis
Since we know that at the used wavelength (Se peak) our Se-MET residues should get a different scattering factor, we could also switch this on with
% refine -m 1o22_truncate-unique_P43212.mtz -p buster_01/refine-coot-0.pdb AutomaticFormfactorCorrection="yes" -d buster_02 | tee buster_02.lis
... let's see who can get the best final model: both in terms of geometry (use e.g. http://molprobity.biochem.duke.edu/ for checking) and R/Rfree. Have a look for missing residues as well; the sequence being
MGSDKIHHHH HHMRLMDILE ILYYKKGKEF GILEKKMKEI FNETGVSLEP VNSELIGRIF LKISVLEEGE EVPSFAIKAL TPKENAVDLP LGDWTDLKNV FVEEIDYLDS YGDMKILSEK NWYKIYVPYS SVKKKNRNEL VEEFMKYFFE SKGWNPGEYT FSVQEIDNLF
After the above refinement run, I would have done two more runs - the first switching on some basic TLS parametrisation (the deposited structure used a much finer segmentation for TLS refinement):
% refine -m 1o22_truncate-unique_P43212.mtz -p buster_02/refine.pdb AutomaticFormfactorCorrection="yes" -M TLSbasic -d buster_03 | tee buster_03.lis
followed by automatic solvent structure update (still using TLS):
% refine -m 1o22_truncate-unique_P43212.mtz -p buster_03/refine.pdb AutomaticFormfactorCorrection="yes" -M TLSalternate -TLS -M WaterUpdatePkmaps -d buster_04 | tee buster_04.lis
With this we get down to an R/Rfree value of 0.1913/0.2285 and the following geometry:
____________________________________________________________________________ | |Clashscore, all atoms: |3.68 |99th percentile* (N=673, 2.04AA +/| |All-Atom|_________________________|______|-_0.25AA)_________________________| |Contacts|Clashscore is the number of serious steric overlaps (> 0.4 AA) per | |________|1000_atoms.________________________________________________________| | |Poor_rotamers____________|4.44%_|Goal:_<1%_________________________| | |Ramachandran_outliers____|0.00%_|Goal:_<0.2%_______________________| | |Ramachandran_favored_____|99.32%|Goal:_>98%________________________| |Protein |Cb_deviations_>0.25AA____|0_____|Goal:_0___________________________| |Geometry|MolProbity score^ |1.65 |93rd percentile* (N=12112, 2.04AA | | |_________________________|______|+/-_0.25AA)_______________________| | |Residues_with_bad_bonds:_|0.00%_|Goal:_0%__________________________| |________|Residues_with_bad_angles:|0.00%_|Goal:_<0.1%_______________________|
You can visualise the trend of R/Rfree for the different runs using
% graph_autobuster_R -d buster_01 -d buster_02 -d buster_03 -d buster_04 -launch
to show something like the below:
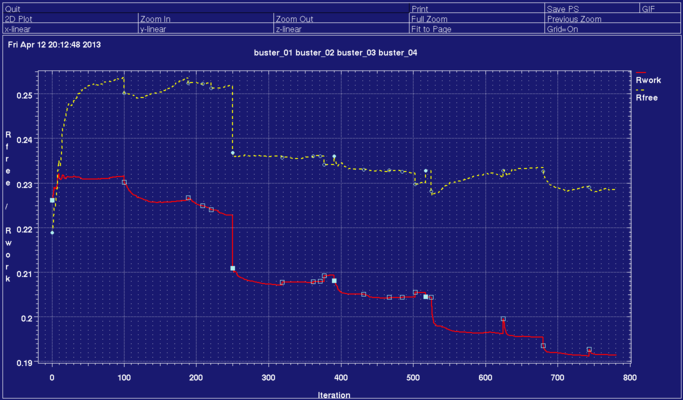
You will need at least two files:
You might also need one or more restraint dictionaries for any bound unusual compound. A good method for generating those is our Grade web-server. Please see the help available there.
If you have a "favourite" deposited PDB file, I can help with the conversion of the deposited structure-factors into a correct reflection file ...
If you have a BUSTER reference card handy and a broswer window pointing to our BUSTER wiki, you should already be in good shape to get started with simple as well as more complicated refinements.
A good quick-start could be using
% refine -p your.pdb -m your.mtz -l your.cif -autoncs -M ShortRun -d buster.01 | tee buster.01.lis
(if you don't have an unusual compound in your model just leave out the "–l your.cif" flag). More complete runs could include
Important: please remember that the workshop computers will take a certain amount of time for running BUSTER - so maybe start with a short and simple example first!
Original page: April 2013, Clemens Vonrhein